311
Радиохимия, 2015, т. 57, N 4, c. 311–320
Органические восстановители ионов Pu и Np в водной технологии
переработки ОЯТ
© В. И. Марченкоа, В. Н. Алексеенкоб, К. Н. Двоеглазов*а
а
Высокотехнологический научно-исследовательский институт неорганических материалов им. акад. А. А. Бочвара,
120360, Москва, ул. Рогова, 5а; * e-mail: KNDvoeglazov@bochvar.ru
б
Федеральный центр ядерной и радиационной безопасности, 119017, Москва, Пыжевский пер., 5
Получено 05.12.2014
УДК 621.039.73
Выполнен обзор научно-технической и патентной информации по применению органических соединений в качестве восстановителей ионов Pu и Np в водной технологии переработки ОЯТ. Установлено,
что органические производные гидразина и гидроксиламина, а также оксимы и производные мочевины
при невысокой кислотности и температуре переводят Np(VI) в Np(V) и Pu(IV) в Pu(III). При увеличении
значений этих параметров происходит медленное восстановление Np(V) до Np(IV). Среди продуктов
окисления органических соединений в азотнокислой среде обнаружены спирты, альдегиды, углеводороды и уксусная кислота. Органические восстановители перечисленных классов могут использоваться на
различных стадиях Пурекс-процесса – для отделения Pu и Np в первом экстракционном цикле, очистки U
от Np во втором цикле очистки урана и реэкстракции Pu на стадии его аффинажа. В лабораторных экспериментах на противоточных экстракционных установках смесителей-отстойников и центробежных аппаратов продемонстрирована возможность достижения высоких коэффициентов разделения U от Pu и/или
Np и глубокого концентрирования плутония c получением реэкстрактов, содержащих до 150 г/л Pu.
Ключевые слова: отработанное ядерное топливо, переработка, экстракция, трибутилфосфат, азотная кислота, уран, плутоний, нептуний, ионы, восстановление, органические соединения.
Введение
Одно из основных направлений развития Пурекспроцесса заключается в минимизации объема жидких
радиоактивных отходов, образующихся в ходе переработки ОЯТ [1]. Для решения этой задачи наряду с
совершенствованием структуры Пурекс-процесса
{например, путем существенного снижения объема
раствора, используемого для растворения топлива [2],
введения операции кристаллизационной очистки урана [3], создания так называемых гибридных технологических схем, сочетающих водные и сухие (пирохимические) операции [4, 5]} представляют интерес
разработки, направленные на максимальное снижение объемов водных рафинатов и реэкстрактов плутония и исключение из состава технологических реагентов солеобразующих соединений. Оба эти пути
непосредственно связаны с поиском альтернативы
редокс-реагентам (их химические свойства и технологические особенности рассмотрены в обзоре [6]),
применяемым в действующей технологии для регулирования валентного состояния ионов Pu и Np.
В качестве восстановителя на ключевой операции
Пурекс-процесса – отделении Pu и Np от U – используется четырехвалентный уран (либо подаваемый
извне в виде азотнокислого раствора, либо генерируемый электрохимически внутри аппарата-реэкстрактора [7]) в смеси с гидразином, а для восстановления
Pu на стадии его аффинажа применяется гидроксиламин, присутствующий в кислых водных растворах в
протонированной форме NH3OH+. Недостатками первого из этих реагентов является присутствие в восстановительном растворе солеобразующего гидразина,
продуктами разложения которого являются HN3 и
NH4NO3, а также значительный избыточный расход
U(IV), обусловленный его окислением азотистой кислотой в фазе экстрагента. К недостаткам гидроксиламина, который в отличие от гидразина является бессолевым реагентом (продукты его разложения – газообразные N2O и N2), следует отнести невысокую скорость реакции с HNO2 и неустойчивость в растворах
HNO3 из-за автокаталитического окисления NH3OH+,
которое ускоряется ионами железа и плутония [8].
Также кинетические особенности реакции гидроксиламина с Pu(IV) {торможение продуктом реакции –
Pu(III) – и сильная обратная зависимость скорости от
кислотности раствора [9]} ограничивают возможности этого реагента по концентрированию Pu и по его
использованию в аппаратах с коротким временем
контакта фаз (центробежные экстракторы, ЦБЭ).
Современная тенденция к повышению выгорания
топлива приводит к усилению перечисленных недостатков U(IV) и гидроксиламина, что делает необходимым поиск других восстановителей Pu и Np – кинетически эффективных, не приводящих к «засолению»
растворов и обладающих свойствами стабилизатораантинитрита. В этой связи интерес представляют органические соединения различных классов, свойства
которых рассматриваются в предлагаемом обзоре.
Замещенные гидразины
Стехиометрия реакций Np(VI) и Pu(IV) с замещенными гидразинами (ЗГ) RN2H4+, где R – органический радикал (как и незамещенный гидразин, ЗГ
присутствуют в кислом растворе в протонированной
312
В. И. Марченко и др.
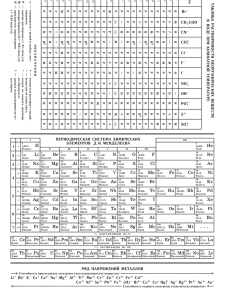
Таблица 1. Константы скорости псевдопервого порядка (k') и время завершения на 99% (τ99) реакций ионов Np и Pu
с замещенными гидразинами при [HNO3] = 1, [Red] = 0.1 моль/л и 25°C
Восстановитель
Гидразин N2H5+
Метилгидразин CH3N2H4+
1,1-Диметилгидразин (CH3)2N2H3+
2-Оксиэтилгидразин HOC2H4N2H4+
Гидразинпропионитрил NCC2H4N2H4+
трет-Бутилгидразин (СH3)3CN2H4+
Фенилгидразин C6H5CH2N2H4+
Карбогидразид N2H3CON2H4+
Окислитель
Np(VI)
Pu(IV)
Np(VI)
Pu(IV)
Np(VI)
Pu(IV)
Np(VI)
Pu(IV)
Np(VI)
Pu(IV)
Np(VI)
Pu(IV)
Np(VI)
Pu(IV)
форме), зависит от соотношения начальных концентраций реагентов – при избытке восстановителя
(Red) происходит перенос двух, а при избытке окислителя (Ox) – четырех электронов [10, 11]. В технологической практике, где обычно используется избыток восстановителя, для большинства ЗГ отношение
стехиометрических коэффициентов Red : Ox = 2, т.е.
на 1 моль Np(VI) или Pu(IV) расходуется 2 моля ЗГ.
Скорость реакций Np(VI) и Pu(IV) с ЗГ описывается кинетическим уравнением (при постоянной ионной силе раствора, μ) [10]
–d[Ox]/dt = k[Ox][Red]/[H+]n,
(1)
в котором порядок относительно ионов H+ (n) равен
или близок к 1. Одинаковая форма кинетических
уравнений для реакций ЗГ с Np(VI) и Pu(IV), а также
тот факт, что скорость большинства из них не зависит от μ, позволили предположить [10], что данные
реакции протекают по одинаковому механизму,
включающему участие в медленной стадии молекулярных (незаряженных) форм ЗГ. Единственным исключением является реакция Pu(IV) с диформилгидразином [N2H2(COH)2], для которой порядок по
Pu(IV) равен 2, а порядок по восстановителю (0.67)
существенно отличается от 1 [12].
Продуктами реакций ЗГ с ионами Np и Pu в азотнокислом растворе, по мнению авторов работы [10],
являются спирты, альдегиды и углеводороды в водном растворе и азот – в газовой фазе. Экспериментально подтверждено образование CH3OH в реакции
Np(VI) с метилгидразином [13] и C2H5OH и
CH3COOH – в реакции Np(VI) с этиловым эфиром
гидразинуксусной кислоты [14].
Восстановленной формой нептуния во всех реакциях Np(VI) с ЗГ при невысокой кислотности и температуре является Np(V); при увеличении этих параметров становится возможным переход Np(V) в
k', мин–1
1.4
7.3·10–4
5.7
4.8·10–3
4.7
2.4·10–3
24.8
0.07
92
0.34
0.42
1.5·10–4
3·102
0.22
τ99, мин
3.3
6.3·103
0.8
9.6·102
0.96
1.9·103
0.18
65.7
0.05
13.5
18.9
3.0·104
0.015
20.9
k'Np/k'Pu
1900
1190
2000
354
215
5300
–
–
Ссылка
[19]
[20]
[13]
[21]
[22]
[23]
[23]
[23]
[24]
[24]
[25]
[26]
[27]
[28]
Np(IV). Реакции Np(V) с ЗГ протекают медленно, но,
как показано на примерах оксиэтилгидразина [15] и
карбогидразида [16], они ускоряются ионами Tc,
концентрация которых в водной фазе на операции
разделения U и Pu в первом цикле Пурекс-процесса
может быть значительной (несколько сотен мг/л).
Соединения из класса ЗГ медленно реагируют с
HNO3 при комнатной температуре (в отсутствие катализаторов) [10]. Количественные данные о скорости этих реакций отсутствуют, за исключением карбогидразида, для которого получено кинетическое
уравнение [17]. По форме оно не отличается от уравнения скорости для реакции HNO3 с незамещенным
гидразином, но скорость окисления карбогидразида
меньше, чем гидразина, и он более устойчив в азотнокислом растворе, в том числе в присутствии ионов
Tc (при [HNO3] ≤ 3 моль/л [18]).
По оценке авторов работ [10, 18], ЗГ быстро взаимодействуют с HNO2 и могут выполнять функцию
стабилизаторов низших валентных состояний актинидов, однако кинетика их реакций с HNO2 не изучена.
В табл. 1 приведены значения констант скорости
псевдопервого порядка k' = k[Red]/[H+]n и рассчитанные с их использованием времена завершения реакций на 99% (τ99) для некоторых ЗГ. Полные данные о
кинетике реакций Pu(IV) и Np(VI) с ЗГ и представителями других классов органических соединений
содержатся в работах [10, 18, 22, 29] и в приводимых
в них ссылках.
Видно, что скорость рассматриваемых реакций
изменяется в широком диапазоне. Так, при [Red] =
0.1 моль/л, [HNO3] = 1 моль/л и 25°C величина τ99
для реакций с Np(VI) изменяется от ~1 с для фенилгидразина до ~20 мин для трет-бутилгидразина, а
для реакций с Pu(IV) – от ~13 мин для гидразинпропионитрила до нескольких десятков часов для
трет-бутилгидразина.
Органические восстановители ионов Pu и Np
Cкорость реакций Np(VI) c большинством ЗГ выше, чем с незамещенным гидразином, но это увеличение относительно невелико и существенно только
в случае оксиэтилгидразина и гидразинпропионитрила. Восстановление Pu(IV) также протекает быстрее,
чем с нитратом гидразина, причем наиболее кинетически эффективными являются гидразинпропионитрил и карбогидразид. Единственным ЗГ, который
взаимодействует и с Np(VI), и с Pu(IV) медленнее,
чем гидразин, является трет-бутилгидразин.
Значения отношения k'Np/k'Pu, приведенные в табл. 1,
показывают, что скорость реакций всех ЗГ с Np(VI)
существенно выше, чем с Pu(IV). Наибольшее различие в скорости (более чем на 3 порядка) отмечается
для 1,1-диметилгидразина (CH3)2N2H4+ и трет-бутилгидразина (CH3)3СN2H4+. Это позволяло ожидать, что
указанные реагенты могут быть использованы для
селективной реэкстракции нептуния в виде Np(V) при
его отделении от U и Pu на операции восстановительной реэкстракции. Однако одностадийные эксперименты с применением этих реагентов в двухфазной
системе, состоящей из 30%-ного ТБФ в додекане, содержащего U(VI), Pu(IV) и Np(VI), и водного раствора
HNO3, показали [23], что одновременно с Np в водную фазу переходит заметное количество Pu [~10 и 2–
3% от исходного в случае (CH3)2N2H4+ и (CH3)3СN2H+4
соответственно] из-за ускоряющего влияния ионов
UO22+ на реакции Pu(IV) c этими ЗГ.
Попытки выполнить селективную реэкстракцию
Np при его отделении от U и Pu на операции восстановительной реэкстракции предпринимались также с
использованием органических соединений других
классов. Японскими исследователями выполнен
цикл работ на противоточных экстракционных установках [30–33] с использованием для этой цели предложенного Колариком [34] н-бутаналя н-C3H7CHO,
результаты которых показали недостаточную полноту реэкстракции Np. Более успешным оказалось применение в качестве реэкстрагента надуксусной кислоты CH3COOOH. В этом случае полнота реэкстракции Np превысила 99% без заметного перехода в
водную фазу U и Pu [35].
Наиболее подробно изученными реагентами из
класса ЗГ являются оксиэтилгидразин и карбогидразид. Оксиэтилгидразин, вероятно, может использоваться для совместной реэкстракции Pu и Np при их
отделении от U в первом экстракционном цикле. Однако для достаточно полной реэкстракции Pu потребуется осуществлять процесс при повышенных значениях температуры и концентрации восстановителя, что может привести к частичному восстановлению Np(V) до экстрагируемого Np(IV) [14] и как
следствие к неполной реэкстракции Np и ухудшению
качества разделения U и Np.
В работе [36] рассмотрена возможность примене-
313
ния оксиэтилгидразина в качестве стабилизатора
Pu(III) вместо традиционного гидразина на операции
восстановительной реэкстракции Pu при отделении
его от U в первом цикле. Помимо солеобразующей
природы гидразина этот вопрос актуален и потому,
что гидразин неустойчив в растворах HNO3 в присутствии ионов Tc [37], и именно это обстоятельство
является основной причиной нарушения процесса
разделения U и Pu [38]. Для торможения каталитического окисления гидразина необходимо по возможности более полно поддерживать технеций в
восстановленном, четырехвалентном, состоянии.
Однако оксиэтилгидразин переводит Tc(VII) в Tc(IV)
очень медленно (на ~60% за 2 ч при [HNO3] = 1,
[HOC2H4N2H4+] = 0.1 моль/л и 40°C) и не может служить полноценной заменой гидразину в качестве
стабилизатора [36].
По-видимому, оптимальным представляется использование оксиэтилгидразина для реэкстракции Pu
на операции его аффинажа, если проводить ее при
повышенной температуре и достаточно высокой концентрации восстановителя. В патенте [39] описан
способ очистки Pu от U, включающий операции совместной экстракции этих элементов из исходного
водного раствора, содержащего до 60 г/л Pu, до
0.6 г/л U и 4.0–4.5 моль/л HNO3, раствором ТБФ в
гексахлорбутадиене, сильно- и слабокислотную промывки экстракта и восстановительную реэкстракцию
Pu раствором 0.8–1.0 моль/л оксиэтилгидразина в
0.2 моль/л HNO3 при температуре от 40 до 60°С и отношении потоков органической и водной фаз О : В =
1.8–2.2. На 15 экстракционных ступенях получен коэффициент очистки Pu от U, равный 50–60, а остаточное содержание Pu в фазе ТБФ составило 10–15 мг/л
при его концентрации в водной фазе ~150 г/л.
Карбогидразид быстро восстанавливает Np(VI)
[16] (кинетика этой реакции подробно не изучена) и
является одним из наиболее быстрых среди ЗГ восстановителей Pu(IV), что позволяет применять его в
ЦБЭ. Одной из особенностей реакции Pu(IV) c карбогидразидом является более сильная, чем для других ЗГ, зависимость скорости от кислотности раствора (порядок относительно HNO3 равен –3 [28]),
вследствие чего его применение для реэкстракции Pu
из раствора ТБФ наиболее эффективно при низкой
кислотности (≤1 моль/л HNO3). Рабочий диапазон
кислотности может быть расширен до ~1.5 моль/л
HNO3 путем введения глицина (аминоуксусной кислоты) СН2(NH2)COOH в состав реэкстрагирующего
раствора [40], что позволяет повысить значение О : В
на операциях реэкстракции Pu.
Возможность глубокого концентрирования Pu
при использовании смеси карбогидразида и глицина
продемонстрирована в опытах, выполненных на растворах облученного топлива ВВЭР-1000 с выгорани-
314
В. И. Марченко и др.
ем 53 ГВт·сут/(т U) после 16-летней выдержки. Во
всех экспериментах экстрагентом служил 30%-ный
ТБФ в разбавителе Isopar-М.
В одном из этих экспериментов проводили восстановительную реэкстракцию Pu при отделении его
от U в условиях первого цикла экстракции на 8-ступенчатом блоке ЦБЭ [28, 41]. Исходный экстракт
имел состав: U 105 г/л, Pu 1.0 г/л, Np 148 мг/л, Tc
134 мг/л. Реэкстрагентом служил водный раствор,
содержащий 0.28 моль/л (NH2NH)2CO, 0.4 моль/л
глицина и 0.2 моль/л HNO3, значение О : В поддерживали равным 12.5. В этих условиях Pu почти полностью переходил в водную фазу (концентрация Pu
в реэкстракте 12.3 г/л при остаточной концентрации
Pu в органическом потоке после этой операции
3.5 мкг/л), а коэффициент очистки U от Pu (KU/Pu)
оценен равным 2.7·105. По расчету также достаточно
полно перешли в водную фазу Np и Tc, при этом
концентрация последнего в реэкстракте составила
1.6 г/л. Высокая полнота реэкстракции Pu при столь
значительной концентрации ионов Tc косвенно указывает на отсутствие существенного каталитического влияния этих ионов на окисление Pu(III) азотной
кислотой и, следовательно, на эффективность карбогидразида как стабилизатора-антинитрита в изученных условиях.
В другом эксперименте восстановительную реэкстракцию Pu из экстракта с содержанием 115 г/л U
и 1.2 г/л Pu проводили в вертикальном аппарате с
насадкой из порошка нержавеющей стали с диаметром зерен 0.4–0.5 мм [42]. В качестве реэкстрагента
использовали раствор, содержащий 0.39 моль/л
(NH2NH)2CO, 0.4 моль/л глицина и 0.2 моль/л HNO3.
Отношение О : В составляло 20, т.е. было в ~2 раза
больше, чем в первом эксперименте. Степень реэкстракции плутония составила 95.3%, а реэкстракт содержал 150 г/л U, 23.5 г/л Pu, 1.7 г/л Np и 1.5 г/л Tc.
При длительной выдержке этого раствора, как и в
первом эксперименте, не наблюдали изменения окраски водного реэкстракта, которое могло бы свидетельствовать об окислении Pu(III).
Высокая полнота реэкстракции Pu получена и при
использовании в качестве восстановителя диформилгидразина [43]. Противоточный эксперимент на
лабораторной установке смесителей-отстойников
(ЭСО) проводили на модельном растворе, содержащем U, Pu, Np и продукты деления, в том числе
Tc, а реэкстрагентом служил раствор 0.5 моль/л
N2H2(COH)2 в 0.2 моль/л HNO3. На 10 ступенях зоны
реэкстракции из органической фазы (30%-ный ТБФ в
н-парафинах) в водную перешло 99.3% Pu.
Замещенные гидроксиламины
Замещенные гидроксиламины (ЗГА) общей формулы RR'NHOH+ (где R = СН3, C3H7 и R' = H, CH3,
C2H5) рассматриваются наряду с ЗГ как перспективные для их использования в технологической практике [10]. Утверждается [10], что среди продуктов
реакций ЗГА с ионами Pu и Np отсутствуют солеобразующие соединения, а конечной формой нептуния
является Np(V).
Стехиометрия реакций ЗГА с Np(VI) и Pu(IV) зависит от соотношения начальных концентраций реагентов. В избытке ионов Np(VI) или Pu(IV) стехиометрический коэффициент Ox : Red равен 6–8 и снижается до (0.5–2) : 1 в избытке восстановителя. По
мнению авторов работы [10], конечными продуктами
реакций ЗГА с Np(VI) и Pu(IV) являются спирт и альдегид. Экспериментально подтверждено образование
формальдегида в реакции Np(VI) с метилгидроксиламином CH3NHOH [44], ацетальдегида и спирта –
в реакции Np(VI) с диэтилгидроксиламином
(C2H5)2NHOH [45] и нитрометана и спирта – в реакции Pu(IV) с (C2H5)2NHOH [при определении состава
продуктов в качестве аналога плутония использовали
Ce(IV)] [46]. В работе [47] отмечается, что при облучении водного раствора 0.2 моль/л диэтилгидроксиламина дозой 10–100 кГр основными продуктами разложения являются газообразные метан, этан, пропан и
бутан. В реакции (C2H5)2NHOH с HNO2 образуются
C2H5OH и CH3CHO при избытке восстановителя и
CH3COOH – при его недостатке [48].
Кинетические уравнения реакций ЗГА с Np(VI)
не отличаются по форме от уравнения (1), установленного для ЗГ, но значение n в случае ЗГА изменяется в более широких пределах (от 0.4 до 1.0). Нецелочисленный порядок по H+-ионам может указывать
на протекание реакций с участием ЗГА по иному,
чем в случае ЗГ, механизму, например по нескольким параллельным путям.
Для реакций ЗГА с Pu(IV) установлены более
сложные кинетические закономерности. Их характерными особенностями являются второй порядок
относительно Pu(IV) и сильное уменьшение скорости с ростом кислотности раствора. Кроме того, реакции с метил-, диметил- и диэтилгидроксиламинами тормозятся продуктом восстановления – Pu(III)
[46, 49, 50]. В общем виде уравнение скорости представляется соотношением
–d[Pu(IV)]/dt = k[Pu(IV)]2[ЗГА]m[Pu(III)]p[H+]n,
(2)
где значения порядков m, n и p для различных ЗГА
находятся в пределах от 1.0 до 2.0, от 2.4 до 4.0 и от
0 до 2 соответственно (при переменной μ порядок
по HNO3 для большинства реакций лежит в интервале 3.3–5.4).
В табл. 2 для реакций Np(VI) и Pu(IV) с отдельными ЗГА приведены значения констант скорости
и τ99 (последние – при [HNO3] = 1, [Np], [Pu] = 0.01,
[Red] = 0.1 моль/л и 25°C).
315
Органические восстановители ионов Pu и Np
Таблица 2. Константы скорости [k, моль/(л·мин) при 25°C] и время завершения на 99% (τ99, мин) реакций ионов Np и
Pu с замещенными гидроксиламинами в водном азотнокислом растворе
Ссылка
Восстановитель
Окислитель
k
τ99
0.12
[51]
Np(VI)
3.8·102
+
Гидроксиламин NH3OH
3.1·103
[9]
Pu(IV)
3.2·10–2
Np(VI)
35.0
0.65
[46]
+
Метилгидроксиламин CH3NH2OH
4.3
[49]
Pu(IV)
2.2·102 а
0.18
[52]
Np(VI)
2.5·102
+
Диметилгидроксиламин (CH3)2NHOH
~0.04
[50]
Pu(IV)
1.7·105
Np(VI)
30.1
1.5
[45]
+
Диэтилгидроксиламин (C2H5)2NHOH
1.6
[46]
Pu(IV)
1.1·105
0.14
[53]
Np(VI)
3.3·102
+
Этилгидроксиэтилгидроксиламин HOC2H4(C2H5)NOH
0.04
[53]
Pu(IV)
7.3·105
а
При 12.5°C.
Из этих данных следует, что восстановление Np(VI)
протекает быстро, особенно в случае HOC2H4(C2H5)·
NOH+ и (CH3)2NHOH+, где реакции завершаются за
время менее 1 мин. Наиболее существенным признаком, отличающим ЗГА как от незамещенного
гидроксиламина, так и от ЗГ, является высокая скорость их взаимодействия не только с Np(VI), но и с
Pu(IV). Как видно из табл. 2, восстановление Pu(IV)
завершается менее чем за 0.1 мин в случае HOC2H4·
(C2H5)NOH+ и (CH3)2NHOH+ и за ~1–1.5 мин при
использовании (C2H5)2NHOH+ и CH3NH2OH+ (в случае NH3OH+ для этого требуется ~3000 мин). Следует отметить, что в производственных растворах скорость некоторых реакций Pu(IV) (в частности, с
CH3NHOH [50] и (C2H5)2NHOH [54]) может быть
еще выше благодаря каталитическому действию ионов железа, присутствующих в этих растворах от коррозии аппаратуры. Авторы работы [54] обращают
внимание на то, что необходим тщательный контроль
за содержанием этих ионов в растворе, поскольку они
могут катализировать также и восстановление Np(V)
до экстрагируемого Np(IV), что снизит полноту реэкстракции Np. В отличие от реакций Pu(IV) с 1,1-диметилгидразином и трет-бутилгидразином, которые
ускоряются ионами UO22+, при восстановлении Np(VI)
диэтилгидроксиламином [55] и Pu(IV) диэтил- [54] и
диметилгидроксиламином [50] не отмечено влияния
U(VI) на скорость реакции при его концентрации до
0.5 моль/л.
Количественные данные об устойчивости ЗГА в
растворах HNO3 отсутствуют. По аналогии с незамещенным гидроксиламином, который автокаталитически окисляется азотной кислотой по схеме [56]
NH3OH+ + 2HNO3 → 3HNO2 + H+ + H2O,
NH3OH+ + HNO2 → N2O + H+ + 2H2O,
(3)
(4)
можно ожидать, что ЗГА при определенных (так называемых «критических») значениях кислотности и
температуры также будут разлагаться по сходному
механизму. Как известно, для NH3OH+ «критические» значения этих параметров снижаются в при-
сутствии ионов Fe и при высокой концентрации Pu
[8]. Не исключено, что такой же эффект может
иметь место и в случае ЗГА.
Большинство исследователей утверждают, что
Pu(III) и Np(V), образующиеся в результате реакций
Pu(IV) и Np(VI) с ЗГА, устойчивы в растворах HNO3
в течение нескольких часов, во всяком случае при
невысокой кислотности и температуре, т.е. ЗГА обладают свойствами стабилизатора-антинитрита. К
сожалению, количественные данные о скорости реакций ЗГА с HNO2 получены только для диэтил- [48]
и диметигидроксиламинов [57] (для последнего – в
хлорнокислом растворе).
Для реакции между (C2H5)2NHOH+ и HNO2 τ99 =
9 с при [HNO3] = 1 моль/л и [(C2H5)2NHOH+] =
0.1 моль/л [48], что в ~2 раза больше, чем для реакции HNO2 с NH3OH+ (в случае гидразина τ99 не превышает 0.01 с при указанных условиях), т.е. диэтилгидроксиламин является недостаточно эффективным
антинитритом.
При электрохимическом исследовании азотнокислых растворов диметилгидроксиламина наблюдали [58] резкое увеличение токов окисления при
[HNO3] > 2 моль/л, обусловленное взаимодействием
(CH3)2NHOH+ с HNO3, вероятно, по схеме, представленной реакциями (3) и (4). Косвенным доказательством недостаточно высокой скорости реакции
(CH3)2NHOH+ с HNO2 является введение в реакционные растворы сульфаминовой кислоты при изучении
реакции Np(VI) с (CH3)2NHOH+ [44] и другого антинитритного реагента – метилгидразина – при исследовании реакции между Pu(IV) и (CH3)2NHOH+ [59,
60]. На основании приведенных данных можно заключить, что вопрос об эффективности замещенных
гидроксиламинов как стабилизаторов, способных
предохранить Np(V) и Pu(III) от окисления азотистой
кислотой, требует дополнительного исследования.
Высокая скорость восстановления Np(VI) и Pu(IV)
замещенными гидроксиламинами позволяет ожидать, что соединения этого класса могут быть ис-
316
пользованы для совместной реэкстракции Pu и Np
при отделении обоих элементов (или каждого из
них) от урана. В патенте [53] на основании расчетных данных о скорости реакций HOC2H4(C2H5)·
NOH+ с Np(VI) и Pu(IV) в водном растворе для этой
цели предлагается применять этилгидроксиэтилгидроксиламин, однако не приводится каких-либо экспериментальных подтверждений его эффективности
при реэкстракции Np и Pu в системах с ТБФ.
Китайские исследователи [61] в рамках разработки технологии APOR (Advanced Purex based on Organic Reductants), ориентированной на использование в Пурекс-процессе органических соединений в
качестве восстановителей ионов Np и Pu [59, 61],
выполнили одностадийные эксперименты по реэкстракции Np(VI) (8.4·10–6 моль/л) из 30%-ного раствора ТБФ в керосине водным раствором 0.05 моль/л
(C2H5)2NHOH+ в 1–2 моль/л HNO3 применительно к
условиям второго уранового цикла. При времени
контакта фаз 5–10 мин и температуре 25°C в водный
раствор перешло более 96% Np.
В работе [59] Pu и U, которые находились первоначально в среде 30%-ного ТБФ в керосине, разделяли с использованием реэкстрагента, содержащего
смесь (CH3)2NHOH+ и метилгидразина. В противоточном эксперименте на 16-ступенчатом блоке ЭСО
после реэкстракции на 10 ступенях (другие 6 ступеней использовались для отмывки реэкстракта Pu от
U) при О : В = 5 KU/Pu составил ~2·104.
Гидроксамовые кислоты
В последние годы внимание исследователей в
качестве потенциальных реагентов для разделения
U, Pu и Np привлекают низшие гидроксамовые кислоты общей формулы RCONHOH, где R = H
(формогидроксамовая кислота, ФГК) и R = CH3
(ацетогидроксамовая кислота, АГК), являющиеся
одновременно и карбоксильными соединениями, и
производными гидроксиламина. Отличительной
особенностью этих реагентов является то, что они
обладают не только восстановительными, но и комплексообразующими свойствами по отношению к
четырехвалентным актинидам [62, 63]. В водном
растворе ФГК и АГК подвергаются кислотному гидролизу, продуктами которого являются гидроксиламин и соответствующая карбоновая кислота
(HCOOH или CH3COOH). Более перспективной для
применения в технологии является АГК, предложенная российскими учеными в 1994 г. [64] и до сих пор
привлекающая внимание исследователей различных
стран [65–69].
Ацетогидроксамовая кислота быстро восстанавливает Np(VI) до Np(V). Подробно кинетика этой
реакции не изучена, установлено только, что ее скорость пропорциональна концентрациям Np(VI) и
В. И. Марченко и др.
АГК [70], а константа скорости второго порядка равна 191.2 л/(моль·с) при 25°C в растворе 1 моль/л
HNO3. Расчет показывает, что в этих условиях реакция завершается за доли секунды даже при невысокой концентрации АГК (0.1 моль/л).
Взаимодействие АГК с Pu(IV) носит сложный
характер, обусловленный тем, что наряду с восстановлением Pu(IV) в системе протекают процессы его
комплексообразования как с самой АГК, так и с продуктом ее гидролиза – уксусной кислотой. Восстановление Pu(IV) происходит медленно, и его скорость уменьшается с ростом кислотности раствора и
концентрации АГК [62, 71]. Первоначально предполагали [72, 73], что непосредственным восстановителем Pu(IV) является другой продукт гидролиза АГК –
гидроксиламин, однако последующие исследования
показали, что, наиболее вероятно, им является сама
АГК [74]. Впоследствии в работе [75] предложен
механизм восстановления Pu(IV), согласно которому
при высоком отношении [АГК] : [Pu] ионы Pu4+ полностью связаны в комплекс с АГК и восстановления
Pu(IV) не происходит (индукционный период на кинетических кривых). При уменьшении этого отношения до ~1 происходит разложение моногидроксаматного комплекса Pu(АГК)3+ по реакции второго
порядка относительно Pu, сопровождающееся восстановлением Pu(IV) до Pu(III), вероятно, путем
внутримолекулярного переноса электрона. При
[АГК] : [Pu] < 1 значительная часть Pu(IV) начинает
переходить в Pu(III) непосредственно по реакции с
АГК, протекающей по кинетическому уравнению
первого порядка относительно реагентов. При определенных условиях авторы работы [60] наблюдали
переокисление образующегося Pu(III) до Pu(IV), которое, по их мнению, происходит, когда концентрации АГК и гидроксиламина близки к нулю. Это явление также отмечали в работе [75], но ее авторы
считают, что оно имеет место и в том случае, когда
АГК еще присутствует в растворе в заметных количествах.
Несмотря на низкую скорость восстановления Pu,
его реэкстракция с использованием АГК протекает
достаточно полно благодаря образованию прочных
гидрофильных комплексов Pu(IV) с АГК [62, 63, 76,
77]. Высокая скорость восстановления Np(VI) этим
реагентом и, очевидно, не менее высокая скорость
его комплексообразования с Pu(IV) и Np(IV) позволяют использовать АГК как для совместной реэкстракции Pu и Np при их отделении от U, так и для
очистки U индивидуально от каждого из этих элементов в ЦБЭ, причем Np первоначально может
присутствовать в органической фазе как в шести-,
так и в четырехвалентном состоянии. В противоточных экспериментах на блоке ЦБЭ [78] при реэкстракции Pu(IV) и Np(IV) водным раствором АГК
(0.2–0.5 моль/л в 0.2–0.5 моль/л HNO3) из 30%-ных
Органические восстановители ионов Pu и Np
растворов ТБФ, содержащих U, получена высокая
степень разделения U и Pu (KU/Pu ~1.5·106). В опытах
по разделению U и Np коэффициент очистки U от
Np (KU/Np) составил 2.15·103 (степень реэкстракции
Np 99.95%).
Авторами работы [79] предпринята попытка применить АГК на операции так называемой «барьерной промывки» для очистки U от остаточных количеств Pu и Np во втором урановом цикле. Из статических опытов следовало, что оптимально реэкстрагент должен содержать 0.2 моль/л HNO3 и 0.3 моль/л
АГК – в этом случае при [U] = 75 г/л в органической
фазе коэффициенты распределения составляют менее 8·10–3 для Np и 2.4·10–4 для Pu. При проведении
этой операции в противоточном эксперименте на
8-ступенчатом блоке ЭСО, когда органическая фаза
содержала U(VI,IV), Pu(IV) и Np(IV), получены невысокие значения KU/Np и KU/Pu (~480 и ~131 соответственно) и, кроме того, отмечалось накопление Np в
зоне отмывки урана «свежим» экстрагентом. Авторы работы [79] подчеркивают, что эти результаты
следует рассматривать как предварительные, поскольку в ходе эксперимента система не достигала
равновесия и не был выдержан оптимальный режим
кислотности.
В создаваемом на Горно-химическом комбинате
(Железногорск Красноярского края) Опытно-демонстрационном центре, предназначенном для отработки технологии переработки высоковыгоревшего
ОЯТ ВВЭР-1000 [80], в качестве базовой принята
одноцикличная экстракционная схема («упрощенный Пурекс»), в которой АГК (в смеси с гидразином) используется на операции совместной реэкстракции Pu и Np с частью урана и на операции барьерной промывки уранового экстракта вместе с
U(IV). В рамках этой схемы может быть реализовано получение реэкстрактов U и Pu, а также их мастер-смесей (вместе с Np или без него). Схема апробирована на стенде смесителей-отстойников в Научно-экспериментальном комплексе Радиевого института при переработке топлива ВВЭР-1000 с выгоранием до 70 ГВт·сут/(т U) [81–83].
Монооксимы
317
от тех, которые установлены для реакций Np(VI) с
ЗГ и ЗГА, а именно они характеризуются первыми
порядками по реагентам и обратной зависимостью
скорости от концентрации HNO3 (H+-ионов). Восстановление Pu(IV) протекает по кинетическому уравнению второго порядка относительно Pu(IV) и характеризуется быстрым начальным падением концентрации Pu(IV) и последующим резким снижением скорости реакции, которое усиливается вследствие торможения ее продуктом – Pu(III).
Скорость восстановления Np(VI) оксимами достаточно велика – реакции с CH3CHNOH и
C3H7CHNOH завершаются за доли минуты. Восстановление Pu(IV) протекает значительно медленнее
(при [HNO3] = 1, [Pu], [Np] = 0.01, [Red] = 0.1 моль/л
и 25°C оно заканчивается за 24 мин в случае
CH3CHNOH и за 140 мин в случае C3H7CHNOH
[87]). При указанных условиях разница в скоростях
реакций Np(VI) и Pu(IV) с CH3CHNOH (~10 раз) и
C3H7CHNOH (700 раз), однако, недостаточно велика
для селективной реэкстракции Np при его отделении
от U и Pu с помощью этих реагентов. В случае бутанальоксима из-за невысокой скорости его реакции с
Pu(IV) совместная реэкстракция Np и Pu при их отделении от U также затруднена, хотя этот процесс
может быть реализован при использовании ацетальдоксима.
В избытке восстановителя продуктами взаимодействия Np(VI) и Pu(IV) с монооксимами в растворе являются масляный альдегид, уксусный альдегид
и ацетон в реакциях с C3H7CHNOH, CH3CHNOH и
(СН3)2СNOH соответственно. При невысокой кислотности и температуре конечной формой Np в реакциях монооксимов c Np(VI) является Np(V). Повышение значений этих параметров приводит к восстановлению Np(V) до Np(IV), которое, однако, протекает с небольшой скоростью.
Устойчивость оксимов в растворах HNO3 наиболее подробно изучена на примере ацетальдоксима
[85]. Его окисление протекает по схеме, аналогичной
представленной уравнениями (3) и (4)
CH3CHNOH + 2HNO3 → 2CH3CHO + 2HNO2 + N2O +
(6)
+ H2O,
CH3CHNOH + HNO2 → CH3CHO + N2O + H2O, (7)
Одно из первых исследований восстановителей
этого класса органических соединений общей формулы R–HC=N–OH выполнено в работе [84], в которой получено кинетическое уравнение реакции между
Np(VI) и ацетоксимом (СН3)2СNOH в хлорнокислой
среде. Позднее в азотнокислом растворе изучены реакции воcстановления Np(VI) ацетальдоксимом
CH3CHNOH [85] и бутанальоксимом C3H7CHNOH
[86].
и ему предшествует индукционный период, продолжительность которого уменьшается с ростом кислотности и/или температуры и не зависит от концентрации Tc(VII) вплоть до 0.02 моль/л. После
окончания индукционного периода наблюдается резкое увеличение концентрации HNO2 в растворе и
быстрое окисление CH3CHNOH.
Кинетические уравнения реакций Np(VI) с
CH3CHNOH и C3H7CHNOH по форме не отличаются
Расчет времени завершения реакции между
CH3CHNOH и HNO2 приводит к величине τ99 ≈ 2 с
318
при [HNO3] = 1, [CH3CHNOH] = 0.1 моль/л и 25°C,
которая значительно больше, чем для традиционного стабилизатора – гидразина. Тем не менее, по мнению авторов работы [88], ацетальдоксим является
достаточно эффективным антинитритом, способным
предохранить Pu(III) от окисления в экстракционных
системах с ТБФ. Их вывод основан на результатах
эксперимента по разделению U и Pu при использовании 30%-ного ТБФ в разбавителе Isopar-М при О : В ≈
7 на 16-ступенчатом блоке лабораторных ЭСО (концентрации обоих металлов в исходной органической
фазе 2 г/л). При использовании в качестве реэкстрагента смеси CH3CHNOH (0.6 моль/л) с NH3ONO3
(0.3 моль/л) в 1.54 моль/л HNO3 потери Pu c органическим потоком составили 0.0046 % (остаточная концентрация Pu в органическом потоке 0.028 мг/л),
тогда как при тех же условиях, но в отсутствие ацетальдоксима, они достигали 12%. Следует отметить,
что в отличие от авторов работы [85], полагающих,
что основным продуктом реакции CH3CHNOH с
HNO2 является уксусный альдегид, авторы работы
[88] считают, что им являются кетоны.
Производные мочевины
В упоминавшемся выше процессе APOR одними
из возможных органических соединений для восстановления Pu и Np рассматриваются производные
мочевины – оксимочевина H2NCONHOH и диоксимочевина HOHNC(O)NHOH. Оба реагента быстро
реагируют с Pu(IV) и Np(VI), очень медленно переводят Np(V) в Np(IV) и одновременно могут выполнять
функции стабилизатора-антинитрита в растворах
HNO3 [89–93]. Разработчики относят эти соединения
к бессолевым восстановителям, несмотря на то что,
по их же данным, одним из продуктов реакции Pu(IV)
с оксимочевиной является нитрат аммония [91].
Результаты статических опытов с обоими восстановителями в двухфазных системах с 30%-ным ТБФ
показали, что полнота реэкстракции Pu уменьшается
с ростом концентрации Pu(IV) и HNO3, а также при
увеличении отношения О : В [89, 91, 93, 94].
Оксимочевину (раствор 0.05 моль/л H2NCONHOH в 1 моль/л HNO3) использовали для реэкстракции Pu из раствора 30% ТБФ в керосине, содержащего 90 г/л U(VI), 46 мг/л Pu(IV) и 52 мг/л Np(VI), в
эксперименте на противоточной 16-ступенчатой установке ЭСО [91] . При О : В = 4 на 8 ступенях реэкстракции значения KU/Pu и KU/Np составили соответственно 4.7·104 и 2.6·102. Поступенчатый пробоотбор в зоне отмывки водного реэкстракта от U обнаружил накопление на ее ступенях Pu и Np в органической фазе, что можно объяснить частичным окислением Pu(III) и Np(V) в этой зоне до экстрагируемых форм – Pu(IV) и Np(VI) – азотистой кислотой.
Реэкстрагент на основе диоксимочевины опробован на нескольких технологических операциях в
В. И. Марченко и др.
противоточных опытах, выполненных с использованием ЭСО.
Отделение Np от U, находящихся в растворе 30%
ТБФ в керосине, испытано на 12-ступенчатом лабораторном блоке, из которых 7 ступеней использовали для реэкстракции Np(VI), а остальные 5 – для отмывки водного потока Np от U [90, 95]. Экстракт
содержал 89 г/л U(VI) и 321 мг/л Np(VI), реэкстрагент – водный раствор 0.05 моль/л HOHNC(O)·
NHOH в 0.5 моль/л HNO3, О : В = 4. В этих условиях
получен KU/Np = 180 (степень реэкстракции нептуния
составила 99.46%), однако, как и в случае оксимочевины, обнаружено накопление Np в зоне отмывки.
Для снижения накопления Np авторы работ [90, 95]
предлагают сократить число ступеней и уменьшить
время пребывания растворов в этой зоне.
Применительно к стадии разделения U и Pu восстановительную реэкстракцию Pu проводили [89] на
16-ступенчатом блоке ЭСО (10 ступеней на реэкстракции и 6 – на отмывке). Экстракт содержал 89 г/л
U(VI) и 247 мг/л Pu, реэкстрагент – водный раствор,
содержащий 0.36 моль/л HNO3 и 0.1 моль/л
HOHNCONHOH, в который было добавлено
0.15 моль/л метилгидразина. При О : В = 4 получен
реэкстракт, содержащий ~1 г/л Pu и 0.65 мг/л U, а
KU/Pu составил 2·104. Авторы работы [89] не наблюдали накопления Pu на ступенях экстрактора, что,
вероятно, является следствием присутствия в составе реэкстрагента эффективного антинитрита – метилгидразина, препятствующего окислению Pu(III) и
Np(V) азотистой кислотой.
Восстановительную реэкстракцию Pu на стадии
аффинажа осуществляли на 12-ступенчатом блоке
ЭСО (8 ступеней на реэкстракции, 4 – на отмывке)
[94]. Исходный экстракт содержал 12 г/л Pu(IV),
реэкстрагентом служил раствор 0.4 моль/л
HOHNCONHOH в 0.4 моль/л HNO3, О : В = 4. Полученный реэкстракт содержал 48 г/л Pu, степень реэкстракции, по оценке авторов, составила 99.96%,
что соответствует остаточному содержанию Pu в
фазе ТБФ около 5 мг/л.
Заключение
Анализ приведенных данных позволяет заключить, что большинство соединений из классов органических производных гидразина, гидроксиламина и
мочевины являются достаточно эффективными восстановителями Np(VI) и Pu(IV) в азотнокислых растворах. При невысокой кислотности и температуре
конечными формами являются Np(V) и Pu(III); при
увеличении значений этих параметров создаются
условия для последующего медленного перехода
Np(V) в Np(IV).
Продуктами окисления органических восстановителей, идентифицированными в реакциях с участием
Органические восстановители ионов Pu и Np
замещенных гидразина и гидроксиламина, являются
спирты и альдегиды в водном растворе, азот и предельные углеводороды в газовой фазе.
На основании имеющихся немногочисленных
экспериментальных данных можно предполагать,
что в отсутствие каталитических примесей наиболее
устойчивыми к окислению в растворах HNO3 являются замещенные гидразины. Однако необходимо
исследование их окисления в растворах, близких по
составу к производственным, в частности содержащих высокие концентрации технеция. Замещенные
гидразины, вероятно, наиболее быстро реагируют с
HNO2, благодаря чему они одновременно могут выполнять функцию стабилизатора низших валентных
состояний Pu и Np. Замещенные гидроксиламины,
оксимы и производные мочевины взаимодействуют
с HNO2 медленнее, и не исключено, что их использование потребует введения в систему кинетически
более эффективного антинитрита, например какоголибо соединения гидразина.
Экспериментами на лабораторных экстракционных установках с использованием в основном модельных растворов продемонстрирована возможность применения упомянутых органических восстановителей на различных стадиях переработки ОЯТ –
отделении Pu и Np от U в первом экстракционном
цикле, очистки U от Np во втором урановом цикле,
реэкстракции Pu на стадии его аффинажа. Применение замещенных гидразинов (карбогидразида и оксиэтилгидразина) на последней операции позволяет
достичь высокой степени концентрирования Pu
(более чем в 20 раз) и получения водных реэкстрактов с содержанием до 150 г/л Pu.
Отдельного изучения требует определение условий безопасного применения рассмотренных органических восстановителей в технологической практике,
поскольку некоторые замещенные гидразины (в частности, диметилгидразин [96]) являются компонентами ракетного топлива или исходными соединениями при получении взрывчатых веществ, а производные гидроксиламина и мочевины способны автокаталитически разлагаться с «пиковым» выделением
большого объема газообразных продуктов [97].
При выборе органического восстановителя с целью использования в технологии переработки ОЯТ
важным этапом представляется проведение его комплексных испытаний на реальных растворах, с обязательным включением операции по утилизации жидких отходов.
Список литературы
[1] Зильберман Б. Я. // Радиохимия. 2000. Т. 42, N 1. С. 3–15.
[2] Федоров Ю. С., Зильберман Б. Я., Алой А. С. и др. //
ЖРХО им. Д. И. Менделеева. 2010. Т. 54, N 3. С. 12–24.
[3] Ohyama K., Nomura K., Washiya T. et al. // Proc. Global’2007.
Boise, Idaho (USA), Sept. 9–13, 2007. P. 1461–1466.
319
[4] Mizuguchi K., Fuse K., Kanamura Sh. et al. // Proc.
Global’2009. Paris (France), Sept. 6–11, 2009. Paper N 9173.
[5] Volk V., Shadrin A., Veselov S. et al. // Proc. Global’2011.
Makuhari (Japan), Dec. 11–16, 2011. Paper N 386756.
[6] Марченко В. И., Двоеглазов К. Н., Волк В. И. // Радиохимия. 2009. Т. 51, N 4. С. 289–302.
[7] Косяков В. Н., Марченко В. И. // Радиохимия. 2008. Т. 50,
N 4. С. 289–300.
[8] Марченко В. И., Журавлева Г. И., Двоеглазов К. Н., Савилова О. А. // Хим. технология. 2007. Т. 8, N 7. С. 318–323.
[9] Колтунов В. С., Журавлева Г. И. // Радиохимия. 1978.
Т. 20, N 1. С. 94–101.
[10] Колтунов В. С., Баранов С. М. // Радиохимия. 1993. Т. 35,
N 6. С. 11–21.
[11] Koltunov V. S., Marchenko V. I. // Proc. RECOD’98. Nice
(France), Oct. 25–28, 1998. Vol. 1. P. 425–431.
[12] Dvoeglazov K., Volk V., Alekseenko V. et al. // Int. Conf.
ATALANTE’2012: Abstracts. Le Corum Montpellier
(France), Sept. 2–7, 2012. P. 213.
[13] Колтунов В. С., Баранов С. М., Жарова Т. П. // Радиохимия. 1987. Т. 29, N 2. С. 155–160.
[14] Баранов С. М., Колтунов В. С., Жарова Т. П. // Радиохимия. 1991. Т. 33, N 2. С. 51–58.
[15] Колтунов В. С., Баранов С. М., Колтунов Г. В. // Радиохимия. 2005. Т. 47, N 2. С. 150–153.
[16] Волк В. И., Марченко В. И., Двоеглазов К. Н. и др. // Радиохимия. 2012. Т. 54, N 2. С. 133–138.
[17] Алексеенко В. Н., Волк В. И., Марченко В. И. и др. // Радиохимия. 2012. Т. 54, N 2. С. 139–142.
[18] Алексеенко В. Н. Карбогидразид: свойства и применение в
водно-экстракционной технологии переработки облученного ядерного топлива: Дис. … к.т.н. М., 2013. 117 с.
[19] Колтунов В. С., Тихонов М. Ф. // Радиохимия. 1973. Т. 15,
N 2. С. 194–199.
[20] Колтунов В. С., Журавлева Г. И. // Радиохимия. 1974.
Т. 16, N 1. С. 84–88.
[21] Колтунов В. С., Баранов С. М., Журавлева Г. И. // Радиохимия. 1989. Т. 31, N 1. С. 50–55.
[22] Taylor R. J., May I., Koltunov V. S. et al. // Radiochim. Acta.
1998. Vol. 81, N 3. P. 149–146.
[23] Баранов С. М., Колтунов В. С. // Радиохимия. 1991. Т. 33,
N 4. С. 58–66.
[24] Колтунов В. С., Баранов С. М., Межов Э. А., Пастущак В. Г. // Радиохимия. 2000. Т. 42, N 2. С.117–120.
[25] Колтунов В. С., Баранов С. М., Жарова Т. П. // Радиохимия. 1993. Т. 35, N 3. С. 25–30.
[26] Колтунов В. С., Пастущак В. Г., Колтунов Г. В. // Радиохимия. 2006. Т. 48, N 4. С. 311–314.
[27] Колтунов В. С., Баранов С. М., Жарова Т. П. // Радиохимия. 1993. Т. 35, N 3. С. 31–38.
[28] Alekseenko V. N., Dvoeglazov K. N., Marchenko V. I. et al. //
J. Radioanal. Nucl. Chem. 2015. Vol. 304, N 1. P. 201–206.
[29] Koltunov V. S. // J. Nucl. Sci. Technol. Suppl. 3. 2002. November. P. 347–350.
[30] Uchiyama G., Hotoku S., Kihara T. et al. // Proc. ISEC’90.
Kyoto (Japan), July 16–21, 1990. Amsterdam: Elsevier, 1992.
Paper N 04-29.
[31] Uchiyama G., Asakura H., Watanabe M. et al. // Proc.
ISEC’96. Melbourne (Australia), 1996. Vol. 2. P. 1291–1296.
[32] Uchiyama G., Asakura H., Hotoku S., Fujine S. // Proc. RECOD’98. Nice (France), Oct. 25–28, 1998. Vol. 1. P. 393–400.
[33] Mineo H., Asakura H., Hotoku S. et al. // Proc. Global’2003.
New Orleans (USA), Nov. 16–20, 2003. P. 1250–1255.
[34] Kolarik Z. US Patent 4659551. 1987.
[35] Зильберман Б. Я., Сытник Л. В., Шадрин А. Ю. и др. Пат.
РФ 2454740 // Б.И. 2012. N 18.
[36] Мелентьев А. Б., Машкин А. Н., Тугарина О. В. и др. //
320
Радиохимия. 2011. Т. 53, N 3. C. 219–224.
[37] Garraway J., Wilson P. // J. Less-Common Met. 1984.
Vol. 97, N 2. P. 191–203.
[38] Машкин А. Н., Беляев Е. М. // Пятая Рос. конф. по радиохимии «Радиохимия-2006»: Тез. докл. Дубна, 23–27 октября 2006 г. Озерск: Маяк, 2006. С. 198–199.
[39] Рамазанов Л. М., Ровный С. И., Глаголенко Ю. В. и др.
Пат. РФ 2307794. Заявл. 12.10.2005. Опубл. 10.10.2007.
[40] Волк В. И., Двоеглазов К. Н., Алексеенко В. Н. и др. Пат.
РФ 2514947. Заявл. 05.03.2012. Опубл. 10.05.2014.
[41] Dvoeglazov K., Alekseenko V., Marchenko V. et al. // Proc.
17th Radiochemical Conf.: Abstracts. Marianske Lazne
(Czech Republic), May 11–16, 2014. P. 201.
[42] Dvoeglazov K., Volk V., Alekseenko V. et al. // Proc.
Global’2013. Salt Lake City (USA), Sept. 29–Oct. 03, 2013.
Paper N 7680.
[43] Двоеглазов К. Н., Логунов М. В., Подрезова Л. Н. и др. //
Седьмая Рос. конф. по радиохимии «Радиохимия-2012»:
Тез. докл. Димитровград, 15–19 окт. 2012 г. С. 122.
[44] Колтунов В. С., Баранов С. М., Жарова Т. П., Шаповалов М. П. // Радиохимия. 1993. Т. 35, N 4. С. 71–78.
[45] Колтунов В. С., Баранов С. М., Жарова Т. П., Абрамина Е. В. // Радиохимия. 1993. Т. 35, N 4. С. 79–84.
[46] Колтунов В. С., Баранов С. М., Шаповалов М. П. // Радиохимия. 1993. Т. 35, N 4. С. 85–92.
[47] Wang J., Bao B., Wu M. et al. // J. Radioanal. Nucl. Chem.
2004. Vol. 262, N 2. P. 451–453.
[48] Zhang A., Hu J., Zhang X., Wang F. // J. Radioanal. Nucl.
Chem. 1998. Vol. 230, N 1–2. P. 235–239.
[49] Колтунов В. С., Баранов С. М. // Радиохимия. 1993. Т. 35,
N 4. С. 54–62.
[50] Chen Y., Tang H., Liu J., He H. // J. Radioanal. Nucl. Chem.
2001. Vol. 289, N 1. P. 41–47.
[51] Колтунов В. С., Тихонов М. Ф. // Радиохимия. 1977. Т. 19,
N 5. С. 611–619.
[52] Колтунов В. С., Баранов С. М., Жарова Т. П., Абрамина Е. В. // Радиохимия. 1993. Т. 35, N 4. С. 49–53.
[53] Baranov S. M., Koltunov V. S., Taylor R. J., May I. US Patent
6444182. Sept. 3, 2002. Appl. July 11, 2000.
[54] Zhang A., Hu J., Zhang X. et al. // J. Radioanal. Nucl. Chem.
2002. Vol. 252, N 3. P. 565–571.
[55] Zhang A., Liu Y. // J. Radioanal. Nucl. Chem. 2000. Vol. 245,
N 2. P. 357–361.
[56] Gowland R., Stedman G. // J. Inorg. Nucl. Chem. 1981.
Vol. 43, N 11. P. 2859–2862.
[57] Li G., He H. // J. Radioanal. Nucl. Chem. 2011. Vol. 287, N 3.
P. 673–678.
[58] Zhang H., Ye G., Cong H. et al. // Proc. Global’2011. Makuhari (Japan), Dec. 11–16, 2011. Paper N 465979.
[59] Liu J., He H., Tang H., Chen Y. // J. Radioanal. Nucl. Chem.
2011. Vol. 288, N 2. P. 351–356.
[60] Hui W., Yan W., Fang L. // Int. Conf. ATALANTE’2012:
Abstracts. Le Corum Montpellier (France), Sept. 2–7, 2012. P. 278.
[61] Zhang A., Hu J., Zhang X., Wang F. // J. Radioanal. Nucl.
Chem. 1998. Vol. 253, N 1. P. 107–113.
[62] Зильберман Б. Я., Федоров Ю. С., Сытник Л. В. и др. //
Хим. технология. 2000. N 6. С. 16–21.
[63] Paulenova A., Tkac P. // Proc. GLOBAL’2007. Boise, Idaho
(USA), Sept. 9–13, 2007. P. 723–727.
[64] Зильберман Б. Я., Машкин А. Н., Нардова А. К. и др. Пат.
РФ 2012075. Заявл. 14.05.1992. Опубл. 30.04.1994.
[65] Guoan Y., Hui H., Wenbin Z. // Int. Conf. ATALANTE’2012:
Abstracts. Le Corum Montpellier (France), Sept. 2–7, 2012. P. 62.
[66] Glatz J.-P., Malmbeck P., Ougier M. et al. // Proc.
Global’2013. Salt Lake City (USA), Sept. 29–Oct. 03, 2013.
Paper N 8196.
[67] Bernier G., Miguirditchain M., Ameil E. et al. // Int. Conf.
ATALANTE’2012: Abstracts. Le Corum Montpellier
В. И. Марченко и др.
(France), Sept. 2–7, 2012. P. 69.
[68] Sukumar S., Pradep Rumar Sharma, Govindan P., Subba
Rao R. V. // J. Radioanal. Nucl. Chem. 2013. Vol. 295.
P. 191–196.
[69] Liu F., Sun Y., Wang H., Zheng W. // J. Radioanal. Nucl.
Chem. 2014. Vol. 299, N 3. P. 1329–1333.
[70] Chung D. Y., Lee E. H. // Proc. Actinide’2005: Final Program and
Abstracts. Manchester (UK), July 4–8, 2005. Paper N 5P26.
[71] May I., Taylor R. J., Wallwork A. L. et al. // J. Alloys Compd.
1998. Vol. 271–273. P. 534–537.
[72] Тaylor R. J., Denniss I. S., May I. // Proc. ATALANTE’2000.
Avignon (France), Oct. 24–26, 2000. Paper N P2-15.
[73] Taylor R. J. // J. Nucl. Sci. Technol. Suppl. 3. November
2002. P. 886–889.
[74] Carrot M. J., Fox O. D., Le Gurun G. et al. // Radiochim.
Acta. 2008. Vol. 96, N 6. P. 333–343.
[75] Tkac P., Precek M., Paulenova A. // Proc. GLOBAL’2009.
Paris (France), Sept. 6–11, 2009. Paper N 9122.
[76] Carrott M. J., Fox O. D., Jones C. J. et al. // Solvent Extr. Ion
Exch. 2007. Vol. 25, N 6. P. 723–745.
[77] Taylor R. J., Sinkov S. I., Choppin G. R., May I. // Solvent
Extr. Ion Exch. 2008. Vol. 26, N 1. P. 41–61.
[78] Birkett J., Carrot M., Fox O. et al. // J. Nucl. Sci. Technol.
2007. Vol. 44, N 3. P. 337–343.
[79] Bernier G., Miguirditchain M., Ameil E. et al. // Procedia
Chem. 2012. Vol. 7. P. 160–165.
[80] Gavrilov P. M., Kudryavtzev E. G., Revenko Yu. A. et al. //
Proc. GLOBAL’2009. Paris (France), Sept. 6–11, 2009. Paper
N 9075.
[81] Gavrilov P. M., Khaperskaya A. V., Fedorov Yu. S. et al. //
Proc. GLOBAL’2011. Makuhari Messe, Chiba (Japan), Dec.
11–16, 2011. Paper N 501241.
[82] Fedorov Yu. S., Zilberman B. Ya., Goletskiy N. D. et al. //
Proc. GLOBAL’2013. Salt Lake City (USA), Sept. 29–Oct.
03, 2013. Paper N 9155.
[83] Голецкий Н. Д., Зильберман Б. Я., Федоров Ю. С. и др. //
Радиохимия. 2014. Т. 56, N 5. C. 427–438.
[84] Шилов В. П., Крот Н. Н., Степанова Е. С. // Радиохимия.
1976. Т. 18, N 2. С. 355–359.
[85] Koltunov V. S., Taylor R. J., Baranov S. M. et al. // Proc.
ATALANTE’2000. Avignon (France), Oct. 24–26, 2000.
Paper N O1-06.
[86] Колтунов В. С., Баранов С. М., Пастущак В. Г. // Радиохимия. 2001. Т. 43, N 4. С. 301–304.
[87] Колтунов В. С., Баранов С. М., Пастущак В. Г. // Радиохимия. 2001. Т. 43, N 4. С. 305–307.
[88] Sze Y.-K., Gosselin J. A. // Nucl. Technol. 1983. Vol. 63, N 3.
P. 431–441.
[89] Yan T., Zheng W., Ye G. et al. // Proc. GLOBAL’2009. Paris
(France), Sept. 6–11, 2009. Paper N 9523.
[90] Yan T. H., Zheng W. F., Zuo C. et al. // Radiochim. Acta.
2010. Vol. 98, N 1. P. 35–38.
[91] Zhu Zh., He J., Zhang Z. et al. // J. Radioanal. Nucl. Chem.
2004. Vol. 260, N 3. P. 601–606.
[92] Zhu Zh., He J., Zhang Z. et al. // J. Radioanal. Nucl. Chem.
2004. Vol. 262, N 3. P. 707–711.
[93] Sivakumar P., Mecnakshi S., Subba Rao R. V. // J. Radioanal.
Nucl. Chem. 2012. Vol. 292, N 2. P. 603–608.
[94] Сянь Лян, Янь Тайхон, Чжен Вейфан и др. // Радиохимия.
2009. Т. 51, N 4. С. 320–322.
[95] Yan T., Zheng W., Ye G. et al. // Proc. Global’2011. Makuhari
(Japan), Dec. 11–16, 2011. Paper N 391241.
[96] Лопырев В. А., Долгушин Г. В., Воронков М. Г. // ЖПХ.
1998. Т. 71, N 8. С. 1233–1248.
[97] Kumar S., Singh P. K., Kamachi Midali U., Natarajan R. // J. Radioanal. Nucl. Chem. 2012. Vol. 292, N 3. P. 1131–1135.