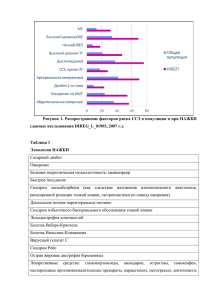
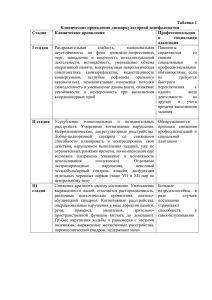
ДЕМЕНЦИЯ ПОЖИЛОГО И СТАРЧЕСКОГО ВОЗРАСТА 1. Деменция - это синдром, который характеризуется потерей памяти в сочетании с невозможностью выполнять работу, ухаживать за собой, совершать простые действия, и деградацией личности. 2. Симптомы слабоумия могут развиться вследствие черепно-мозговой травмы, инфекционных заболеваний центральной нервной системы, интоксикаций, последствий перенесенного инсульта, болезни Альцгеймера или паркинсонизма. Вторичное поражение головного мозга развивается у пациентов с патологией церебральных сосудов. 3. Обычно слабоумие развивается у пожилых людей, но первые признаки заболевания могут возникнуть и в раннем возрасте. В этом случае причиной развития деменции может быть наследственная предрасположенность, объёмные образования головного мозга, артериальная гипертензия, алкогольная болезнь, употребление наркотических веществ. Симптомы слабоумия возникают у лиц, имеющих низкий социальный статус. 4. При старческой деменции нейроны погибают постепенно. По этой причине не происходит резкого снижения интеллектуальных и психических функций. В дебюте заболевания у людей снижается память, внимание, способность учиться. Это часто воспринимается как признак переутомления или симптом других заболеваний нервной системы. 5. Деменция характеризуется нарушением познавательных функций; нарушениями ориентации во времени, месте и в рамках собственной личности; расстройствами в поведении; нарушениями мышления; понижением критического отношения к себе и окружению; эмоциональными расстройствами; расстройствами восприятия. 6. Факторы, которые способны спровоцировать и ускорить развитие деменции: неправильный образ жизни; сахарный диабет; хронические инфекционные заболевания; проведенный ранее гемодиализ; почечная и печеночная недостаточность больного; патологии сосудистой системы; хронические заболевания внутренних органов; перенесенные черепно-мозговые травмы; сопутствующие заболевания (болезнь Альцгеймера, Паркинсона, Пика). 7. В группу риска попадают люди возрастом старше 65 лет, страдающие от повышенного давления, имеющие повышенное содержание липидов в крови, с лишним весом и недостатком физической активности, также недостаток умственной деятельности больного на протяжении длительного времени и сниженное содержание гормона эстрогена у женщин. 8. Полное восстановление клеток мозга невозможно, но ранняя диагностика деменции у пожилых людей позволит не усугубить ситуацию. Поэтому своевременный визит к доктору позволит начать лечение как можно раньше. При обнаружении признаков болезни нужно обратиться к терапевту, который даст направление к узкоспециализированному специалисту - неврологу, психиатру, гериатру (врачу по болезням пожилого возраста). 9. Диагностика старческой деменции включает оценку когнитивных способностей, ЭЭГ, компьютерную томографию и ряд неврологических исследований. На основе полученных результатов врач может дать прогнозы деменции и назначить терапию, замедляющую развитие заболевания. 10. Существуют современные методы лечения деменции, основанные на изучении спинномозговой жидкости и данных, полученных в результате позитронно-эмиссионной томографии. Генная терапия направлена на предотвращение отложений белков в головном мозге, которые провоцируют дегенеративные процессы и ухудшение состояния больного. Ученые ведут разработку вакцин от слабоумия и старческой деменции. СИНДРОМ МАРТИНА-БЕЛЛ (синдром ломкой X-хромосомы) Автор, описавший синдром В 1943 году физиологи из Великобритании Д. Мартин и Д. Белл изучали 11 случаев олигофрении у мужчин из одной семьи, в которой женщины имели нормальное интеллектуальное развитие. Генетическая основа заболевания была выявлена в 1969 году американским генетиком Г. Лабсом. Частота встречаемости Распространенность среди мальчиков составляет 1:4000, среди девочек – 1:6000. Этиология Синдром Мартина-Белл является результатом дефекта гена FMR1, расположенного в Xхромосоме. Наследование происходит по доминантному сцепленному с полом типу с неполной пенетрантностью. У мужчин присутствует одна X-хромосома, поэтому мутантный аллель всегда провоцирует болезнь. У женщин есть две половые хромосомы типа X: одна активная, другая – резервная, инактивированная. Таким образом, при наличии мутации в одном из двух генов FMR1 заболевание проявляется или нет в зависимости от активности измененной хромосомы. Мужчины с ломкой хромосомой X не могут передать ее сыновьям, но передают всем дочерям, которые либо болеют, либо остаются здоровыми носителями мутации. Женщины с дефектной хромосомой передают ее детям обоих полов с вероятностью 50%. Наследование синдрома учащается от поколения к поколению, этот феномен называется парадоксом Шермана. Характерные клинико-психологические особенности Дети рождаются с увеличенной массой тела, в среднем – 3,5-4 кг. Характерен макроорхизм – увеличение яичек без эндокринного заболевания. Окружность головы больше нормы или соответствует ее верхним границам. Лоб высокий и широкий, лицо вытянутое с уплощенной средней частью. Нос имеет слегка клювовидный загиб, ушные раковины крупные, располагаются низко. Суставы отличаются хорошей подвижностью, кости кистей и стоп широкие. Кожа зачастую гиперэластичная, волосы и радужные оболочки глаз светлого оттенка. Своеобразие речи: ускоренная, сбивчивая, изобилует повторами, эхолалиями и персеверациями. Аутистические расстройства представлены трудностями коммуникации и поведенческими нарушениями. Дети проявляют агрессивность и замкнутость при попытке установления контакта. В тяжелых случаях развивается мутизм полное отсутствие речи как средства общения. В поведении преобладает двигательная расторможенность, гиперактивность, стереотипии, самоповреждения. Пациенты избегают смотреть в глаза, не допускают прикосновений, но по сравнению с больными аутизмом интерес к общению присутствует. Стереотипные движения включают хлопки руками, прыжки, вращения вокруг своей оси, встряхивания руками, бег по кругу, гримасничанье и однообразное хныканье. Имеются трудности планирования и контроля поведения, переключения внимания и пространственной координации. Определяется легкое снижение мышечного тонуса, двигательная дискоординация. У части больных имеются глазодвигательные нарушения, усиление сухожильных рефлексов, экстрапирамидные паракинезы. При тяжелых формах синдрома возникают эпилептические припадки. У 25% пациенток с премутационным состоянием развивается первичная недостаточность яичников. Особенности интеллектуального развития Умственная отсталость лёгкой или средней степени тяжести. СИНДРОМ КОРНЕЛИИ ДЕ ЛАНГЕ Автор, описавший синдром В 1933 году голландский педиатр Корнелия де Ланге впервые подробно описала проявления данного синдрома у 5 детей. В 1916 году немецкий доктор Брахман В. описал аналогичного пациента. Частота встречаемости Частота заболевания 1:10000. Встречается на всех континентах у людей всех рас и этнических групп с одинаковой частотой для обоих полов. Этиология Примерно половина случаев обусловлена мутациями в гене NIPBL, около 5 % случаев мутациями в гене SMC1A, кодирующем субъединицу белкового комплекса когезина. Один описанный случай связан с мутацией в гене SMC3, который также кодирует одну из субъединиц когезина. В ряде случаев при цитогенетическом исследовании находят микродупликацию локусов q25 - q29 хромосомы 3. Однако причины, вызывающие эти генные мутации, пока находятся в стадии теорий и предположений. Есть мнение, что их могут вызывать инфекционные заболевания беременной, особенно в первом триместре, некоторые лекарственные препараты, преклонный возраст отца ребенка либо возраст рожениц старше 35 лет, а также браки между родственниками. Но, ни одна из перечисленных причин не является бесспорной и 100 % вызывающей генетические изменения. Еще прорабатывается гипотеза, что синдром Корнелии де Ланге передается по наследству, однако больше, чем у половины обследованных пациентов это заболевание возникло спорадически, впервые в роду. Характерные клинико-психологические особенности Микроцефалия; брахицефалия; тонкие сросшиеся брови; длинные загнутые ресницы; деформированные ушные раковины; маленький нос, открытые вперед ноздри, атрезия хоан; тонкая верхняя губа; микрогения; высокое нёбо или расщелина нёба; нарушение прорезывания зубов; миопия, косоглазие, астигматизм, атрофия зрительных нервов, колобома зрительного нерва; маленькие кисти и стопы, отсутствие или значительное недоразвитие проксимальных отделов конечностей, вследствие чего кисти и стопы кажутся прикрепленными непосредственно к туловищу, уменьшение количества пальцев; мраморная кожа; гипоплазия сосков; гипертрихоз; судороги; врожденные пороки внутренних органов (сердца, почек, пилоростеноз, крипторхизм); аутизм или аутистическое поведение. У части больных наблюдается бег по кругу, стереотипные движения руками и самоповреждения. У всех больных отмечается отставание в росте и весе, типичны рецидивирующие респираторные инфекции. Выделяют два варианта синдрома: первый (классический) с выраженной пренатальной гипоплазией, значительной задержкой физического и интеллектуального развития, грубыми пороками развития; второй - с аналогичными лицевыми и малыми скелетными аномалиями, но пограничной задержкой психомоторного развития и отсутствием грубых пороков развития. Особенности интеллектуального развития В 80 % случаев тяжёлая умственная отсталость (имбецильность), в оставшихся 20 % менее выраженные интеллектуальные нарушения. ГИДРОЦЕФАЛИЯ Автор, описавший синдром В древней медицинской литературе гидроцефалия описывалась редко, хотя её существование и симптоматика были хорошо известны. Отец медицины Гиппократ считается первым врачом, попытавшимся задокументировать лечение гидроцефалии. Эвакуация поверхностной внутричерепной жидкости у детей с гидроцефалией была впервые подробно описана Ибн Синой. Частота встречаемости 1-2 случая на 1000 новорожденных. Этиология К гидроцефалии могут приводить три патогенетически различных механизма: повышение продукции цереброспинальной жидкости, механические препятствия на пути ее циркуляции и нарушение ее всасывания. Особенно часто гидроцефалия является результатом постнатальных менингоэнцефалитов и черепно-мозговых травм, реже она развивается внутриутробно. Известны также формы гидроцефалии, вызываемые менделирующими генами, как аутосомно-рецессивным, так и рецессивным, локализованным в Х-хромосоме. Характерные клинико-психологические особенности Увеличенный размер головы и изменения на рентгеновском снимке: истончение костей черепа, расхождение швов, усиление пальцевых вдавлений, расширение кожных вен на голове. Часто отмечаются экзофтальм с нарушением движения глазных яблок вверх, нистагм, косоглазие; нередко гидроцефалии сопутствуют неврологические изменения: параличи, парезы, повышение сухожильных рефлексов; на глазном дне часто обнаруживаются застойные явления и вторичная атрофия зрительных нервов. У некоторых больных поражается слух и возникают вестибулярные расстройства. В ряде случаев бывают судорожные приступы. При окклюзионной гидроцефалии могут возникать пароксизмальные кризы с резкой головной болью, рвотой, выраженными вегетативными нарушениями. В более тяжелых случаях возникают симптомы поражения ствола, нарушения дыхания, тонические судороги, расстройства глазодвигательной иннервации. Особенности интеллектуального развития Степень умственной отсталости при гидроцефалии, как правило, пропорциональна степени самой гидроцефалии и ее тяжелого следствия - истончения мозговой коры. Интеллектуальные нарушения при приобретенной гидроцефалии обычно более глубокие, чем при врожденных формах, сочетающихся с уродствами. Это зависит от сопутствующих изменений паренхимы мозга, которые отмечаются в случаях приобретенной гидроцефалии, возникшей вследствие менингоэнцефалитов, травм и т. п. МИКРОЦЕФАЛИЯ Автор, описавший синдром Микроцефалия как отдельная форма олигофрении была описана в XIX веке (И. П. Мержеевский, 1871; Д. С. Зернов, 1879; С. С. Корсаков, 1894 и др.). Частота встречаемости Частота встречаемости порока колеблется приблизительно от 1:2000 до 1:10000 среди всех новорождённых детей. Как самостоятельное заболевание истинная микроцефалия встречается гораздо реже и выявляется у 1 из 50000 детей как у девочек, так и у мальчиков. Если же данная патология возникает как проявление другой наследственной болезни, то её частота в популяции обуславливается частотой основного заболевания. Этиология Первичная микроцефалия является компонентом наследственных болезней с аутосомнорецессивным и рецессивным, сцепленным с полом типами наследования (синдром Джакомини, синдром Пейна). Вторичная микроцефалия отмечается при хромосомных аберрациях, наследственных энзимопатиях (фенилкетонурии), патологии беременности и родов. Вторичная эмбриопатическая микроцефалия обусловлена воздействием на плод тератогенных факторов и может являться следствием внутриутробных инфекций и интоксикаций, радиационного влияния, гипоксии, внутричерепных родовых травм, метаболических нарушений, гормональных заболеваний матери. Характерные клинико-психологические особенности Симметричное уменьшение размера мозгового черепа при нормальных или незначительно уменьшенных размерах лица. Лоб, как правило, уплощен, отмечаются увеличенные ушные раковины, удлиненный нос, косоглазие. Хотя другой соматоневрологической симптоматики обычно нет, все же описаны различные семьи со своеобразными формами микроцефалии, сочетающейся с разными дополнительными симптомами (поражение глаз, почек, своеобразное строение лица, конечностей и т. д.) При вторичной микроцефалии при нерезком уменьшении размеров черепа деформации его могут носить более грубый характер. Нет равномерности в уменьшении мозговой части черепа, затылок, как правило, уплощен. Отмечается более глубокое психическое недоразвитие, которое нередко сочетается с судорожными припадками и другими дополнительными симптомами и синдромами, а также с очаговыми неврологическими симптомами. У людей с данным заболеванием чаще встречается отставание в росте и массе тела, диспропорциональность телосложения. Особенности интеллектуального развития Умственная отсталость постоянно сопутствует микроцефалии. Обычно она имеет равномерный характер и достигает глубоких степеней. Наряду с тяжелыми формами слабоумия нередко встречается олигофрения в степени дебильности. ФЕНИЛКЕТОНУРИЯ Автор, описавший синдром Фенилкетонурия была открыта в 1934 году норвежским врачом Иваром Фёлингом, обратившим внимание на повышенную концентрацию продуктов распада фенилаланина в моче у детей с задержкой умственного развития. Частота встречаемости Распространённость фенилкетонурии широко варьируется во всём мире. В Европе частота встречаемости данного заболевания составляет 1 случай на 10 000 живорождённых детей, но для некоторых районов она значительно выше, что связано с большим количеством близкородственных браков. Этиология Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена. Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Характерные клинико-психологические особенности Специфический неприятный запах изо рта, от кожи или мочи, который можно сравнить с затхлым "мышиным" запахом; приступы тошноты и рвоты; светочувствительность; неврологические нарушения, которые нередко могут включать эпилепсию (50 %); экстрапирамидные нарушения (паркинсонизм — неврологический синдром, который характеризуется тремором в состоянии покоя, мышечной ригидностью и общей замедленностью движений); нарушения зрения: косоглазие, атрофия зрительного нерва, снижение остроты зрения; кожные высыпания по типу экземы, локализующиеся на любых участках тела; светлая кожа и голубые глаза (из-за невозможности фенилаланина превращаться в меланин); микроцефалия; гиперактивность: беспокойство, неконтролируемые вспышки гнева, невозможность сосредоточиться на выполняемых задачах, спонтанное снижение концентрации; интеллектуальная недееспособность: от лёгкой недостаточности интеллекта до идиотии и имбецильности; проблемы в поведенческой, эмоциональной и социальной сферах: трудности обучения в образовательных учреждениях, снижение мотивации при выполнении рабочих задач, снижение социальных коммуникаций, раздражительность, перепады настроения, социальная изоляция; психические расстройства: депрессии, генерализованные тревоги, шизофрения, биполярное расстройство. У детей, рождённых от матерей с фенилкетонурией и неконтролируемым уровнем фенилаланина в крови, риск умственной отсталости значительно выше, поскольку они подвергаются воздействию очень высоких и токсичных уровней фенилаланина ещё до рождения. Такие дети могут иметь низкий вес при рождении, что вызывает проблемы прибавки массы и физического развития в будущем. Кроме того, умственно они развиваются значительно медленнее, чем их здоровые сверстники. Другими характерными признаками являются различные пороки сердечно-сосудистой системы (тетрада Фалло, коарктация аорты, дефект межжелудочковой перегородки, дефект межпредсердной перегородки) и поведенческие проблемы, связанные в будущем с нарушением социальной адаптации. Особенности интеллектуального развития Диагноз олигофрения может переходить в задержку психического развития (до 3 лет) и даже норму, что совсем нехарактерно для других разновидностей олигофрений. СИНДРОМ ГУРЛЕРА Автор, описавший синдром Заболевание впервые было описано австрийским врачом-педиатром Гертрудой Гурлер. Частота встречаемости 1 случай на 100 тысяч живых новорожденных. Мальчики и девочки болеют одинаково часто. Этиология Возникновение заболевания связано гетерогенными мутациями (мелкими делециями, дефектами сайта сплайсинга) гена IDUA, кодирующего синтез энзима альфа-Lидуронидазы. Ген локализован в коротком плече 4 хромосомы в локусе 4p16.3. Наиболее частые мутации гена - Q70X и W402X. Лизосомальный фермент α-L-идуронидаза регулирует метаболизм основных структурных компонентов межклеточного матрикса соединительной ткани - отвечает за деградацию гликозаминогликанов гепарансульфата и дерматансульфата. Мукополисахаридоз типа I наследуется по аутосомно-рецессивному типу. Это означает, что больной ребенок может родиться только в случае, если оба родителя являются носителями болезни, имея по одной копии нормального гена и по одной копии «дефектного» гена (такие носители клинически здоровы). Тогда с вероятностью 25% ребенок получит от обоих родителей именно «дефектный» ген, и в этом случае возникает болезнь. Характерные клинико-психологические особенности Увеличенная голова необычной формы, грубые черты лица, большие лобные бугры, запавшая переносица, широкий кончик носа, толстые губы, приоткрытый рот, увеличенный язык, короткая шея, низкий рост. Пальцы короткие и плохо гнутся, ограничена подвижность также и в других суставах, из-за чего дети начинают ходить позже сверстников и на полусогнутых ногах. Часто у больных диагностируют дисплазию тазобедренных суставов. Грудная клетка деформирована. Живот увеличен из-за большого размера печени и селезенки. Возникают изменения глаз (включая помутнение роговицы), ухудшаются зрение и слух. Со стороны сердечно-сосудистой системы обычны пороки сердечных клапанов. Из-за воздействия болезни на дыхательные пути возникают частые респираторные инфекции. Особенности интеллектуального развития Сначала интеллектуальное развитие происходит по возрасту, но уже к 1-1.5 годам замедляется, а к 2-4 годам постепенно прекращается. Затем возникает регресс, то есть утрата уже приобретенных навыков. УМСТВЕННАЯ ОТСТАЛОСТЬ ПРИ ГЕМОЛИТИЧЕСКОЙ БОЛЕЗНИ НОВОРОЖДЕННЫХ Частота встречаемости 1:250-300 родов Этиология Гемолитическая болезнь новорожденных (эритробластоз плода) возникает при иммунологической несовместимости крови матери и плода, чаще у ребенка с группой крови А (II) или В (III) при наличии у матери 0 (I) группы крови, а чаще всего - при несовместимости по резус-фактору. Заболевание развивается у резус-положительного ребенка в том случае, если мать имеет отрицательный резус-фактор. Для появления конфликта имеют значение повышенная чувствительность организма матери к сенсибилизации резус-фактором, состояние эндокринной системы, повышенная проницаемость сосудов, в особенности сосудов плаценты. Вероятность возникновения и появления тяжелых форм гемолитической болезни новорожденных увеличивается с каждой последующей беременностью вследствие сенсибилизации организма матери. Факторами, способствующими возникновению гемолитической болезни, являются аборты, переливание резус-положительной крови, повреждение плаценты. Характерные клинико-психологические особенности Экстрапирамидные двигательные расстройства, дисплазия зубной эмали, выпадение зубов, дефекты слуха, умственная отсталость, глазодвигательные расстройства, опистотонус, судороги, гиперкинетический синдром, атаксия, дистония мышц конечностей, парезы, параличи. Отмечается чувствительность к тактильным раздражителям, низкий болевой порог, повышенная эмоциональная возбудимость, эгоцентризм, слабая работоспособность. Настроение неустойчивое; наблюдаются быстрые переходы от спокойного состояния к бурным аффектам, злобности, агрессии. Особенности интеллектуального развития Интеллектуальная недостаточность у разных больных может варьировать от легкой дебильности до идиотии и обычно характеризуется своеобразным дисгармоническим недоразвитием отдельных психических функций.