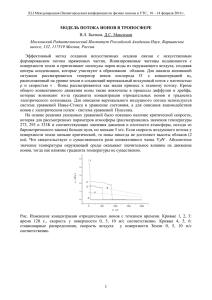
1. Предмет электрохимии. Проводники первого и второго рода. ПРЕДМЕТ ЭЛЕКТРОХИМИИ. Электрохимия является разделом физической химии, в котором изучаются законы взаимосвязи химических и электрических явлений. Основным предметом электрохимии являются процессы, протекающие на электродах при прохождении тока через растворы, электродные процессы. Можно выделить два основных раздела электрохимии: термодинамику электродных процессов, охватывающую равновесные состояния систем электрод - раствор, и кинетику электродных процессов, изучающую законы протекания этих процессов во времени. Электрохимия изучает также теорию электролитов. Электрохимия имеет очень большое значение, т.к. закономерности электрохимии являются теоретической основой для разработки важных технических процессов электролиза и электросинтеза, т.е. получения химических продуктов на электродах при прохождении тока через растворы (получение хлора и щелочей, получение и очистка цветных и редких металлов, электросинтез органических соединений). Важной областью практического применения электролиза является гальванотехника электропокрытие металлами. Другая важная область техники, в основе которой лежат электрохимические процессы, это создание химических источников тока (гальванических элементов, в том числе аккумуляторов), в которых химическая реакция используется как источник электрического тока. Большое развитие получили электрохимические методы химического анализа (электроанализ, кондуктометрия, потенциометрия, полярография и др.). Возникновение электрохимии как науки связано с именами Гальвани, Вольта и Петрова, которые на рубеже XVIII и XIX в. открыли и исследовали электрохимические (гальванические) элементы. Деви и Фарадей в первой половине XIX в. изучали электролиз. Быстрое развитие электрохимии в конце XIX в. связано с появлением теории электролитической диссоциации Аррениуса (1887) и с работами Нернста по термодинамике электродных процессов. Теория Аррениуса развита Дебаем и Гюккелем (1923), которые разработали электростатическую теорию. Для последних десятилетий характерно быстрое развитие электрохимической кинетики, изучение явлений перенапряжения, коррозии, гальванических покрытий и др. ПРОВОДНИКИ ПЕРВОГО И ВТОРОГО РОДА. При наложении электрического поля в проводниках происходит перемещение свободных зарядов. Проводники первого рода – это проводники, в которых носителями зарядов являются электроны. Например, металлы в твердом и в жидком состояниях. В проводниках второго рода носителями зарядов являются положительные или отрицательные ионы. Это электролиты – расплавы оксидов, гидроксидов металлов, солей или водные растворы солей. Твердые и жидкие проводники, прохождение через которые электрического тока не вызывает переноса вещества в виде ионов, называются проводниками первого рода. Электрический ток в проводниках первого рода осуществляется потоком электронов (электронная проводимость). К таким проводникам относятся твёрдые и жидкие металлы и некоторые неметаллы (графит, сульфиды цинка и свинца). Вещества, прохождение через которые электрического тока вызывает передвижение вещества в виде ионов (ионная проводимость) и химические превращения в местах входа и выхода тока (электрохимические реакции), называются проводниками второго рода. Типичными проводниками второго рода являются растворы солей, кислот и оснований в воде и некоторых других растворителях, расплавленные соли и некоторые твёрдые соли. Как правило, в проводниках второго рода электричество переносится положительными (катионы) и отрицательными (анионы) ионами, однако некоторые твёрдые соли характеризуются униполярной проводимостью, то есть переносчиками тока в них являются ионы только одного знака катионы (например, в AgCl) или анионы (BaCl2, ZrO2+CaO, растворы щёлочных металлов в жидком аммиаке). Деление проводников в зависимости от типа проводимости (электронная или ионная) является условным. Известны твёрдые вещества со смешанной проводимостью, например Ag2S, ZnO, Cu2O и др. В некоторых солях при нагревании наблюдается переход от ионной проводимости к смешанной (CuCl). Проводники второго рода называются электролитами. Это могут быть чистые вещества или растворы. Часто электролитами называют вещества, растворы которых проводят электрический ток. Эти растворы называют растворами электролитов. 2. Электрохимические реакции. Законы Фарадея. ЭЛЕКТРОХИМИЧЕСКИЕ РЕАКЦИИ. Электрохимические реакции протекают на границе электрод (проводник первого рода) электролит (проводник второго рода). Они вызваны невозможностью для электронов носителей тока в электродах свободно двигаться в электролите. Эти реакции состоят в обмене электронами между электродом и ионами (молекулами) в растворе. На катоде электроны переходят от электрода к иону (или молекуле), на аноде от иона (молекулы) к электроду, при этом ионы (молекулы) теряют или изменяют свой электрический заряд. Это первичная электрохимическая реакция, продукты которой нередко вступают в дальнейшие реакции, не связанные непосредственно с переносом тока ионами. Примерами катодных реакций могут служить следующие реакции: Cu2+ + 2e Cu (1) Fe3+ + e Fe2+ (2) 2H3O+ + 2e H2(г) + 2H2O (3) На аноде могут протекать реакции типа: 4OH- O2 + 2H2O + 4e (4) Fe2+ Fe3+ + e (5) 2Cl- Cl2(г) + 2e (6) Zn Zn2+ + 2e (7) Материал электрода может участвовать в электрохимической реакции реакция (7), но может быть и инертным (остальные реакции). В последнем случае на поверхности электрода могут выделяться металлы реакция (1) или газы реакции (3, 4, 6). Наконец, электрохимическая реакция может протекать и при отсутствии перехода ионов из раствора к электроду и обратно. В этих случаях перенос электричества осуществляют только электроны, но у поверхности электрода в растворе ионы изменяют свою валентность реакции (2) и (5). Совокупность двух электрохимических реакций, из которых одна протекает на катоде, а другая на аноде, даёт химическую реакцию электролиза или реакцию, протекающую в электрохимическом элементе: или Cu2+ + 2Cl- Cu(т) + Cl2 (1) + (6) 2H3O+ + 2OH- 1/2 O2 + H2 + 3H2O (3) + 1/2 (4) H2O 1/2 O2 + H2 ЗАКОНЫ ЭЛЕКТРОЛИЗА (ЗАКОНЫ ФАРАДЕЯ). Поскольку прохождение электрического тока через электрохимические системы связано с химическими превращениями, между количеством протекающего электричества и количеством прореагировавших веществ должна существовать определенная зависимость. Она была открыта Фарадеем и получила свое выражение в первых количественных законах электрохимии, названных впоследствии законами Фарадея. Первый закон Фарадея. Количества веществ, превращённых при электролизе, пропорциональны количеству электричества, прошедшего через электролит. m = kэIt = kэq m – количество прореагировавшего вещества; kэ – некоторый коэффициент пропорциональности; q – количество электричества, равное произведению силы тока I на время t. Если q = It = 1, то m = kэ, т.е. коэффициент kэ представляет собой количество вещества, прореагировавшего в результате протекания единицы количества электричества. Коэффициент kэ называется электрохимическим эквивалентом. Второй закон Фарадея отражает связь, существующую между количеством прореагировавшего вещества и его природой: при постоянном количестве прошедшего электричества массы различных веществ, испытывающие превращение у электродов (выделение из раствора, изменение валентности), пропорциональны химическим эквивалентам Ai этих веществ: mi /Ai = const Обобщенный закон можно представить в виде такой формулировки: отношение массы полученного вещества к его химическому эквиваленту равно отношению использованного заряда к тому, который надо потратить на извлечение одного моля вещества. m/A = q/F. Можно объединить оба закона Фарадея в виде одного общего закона: для выделения или превращения с помощью тока 1 г-экв любого вещества (1/z моля вещества) необходимо всегда одно и то же количество электричества, называемое числом Фарадея (или фарадеем). m = A M It = It F zF Точно измеренное значение числа Фарадея F = 96484,52 0,038 Кл / г-экв (Кл/моль) Таков заряд, несомый одним грамм-эквивалентом ионов любого вида. Умножив это число на z (число элементарных зарядов иона), получим количество электричества, которое несёт 1 г-ион. Разделив число Фарадея на число Авогадро, получим заряд одного одновалентного иона, равный заряду электрона: e = 96484,52 / (6,0220351023) = 1,602191310-19 Кл Законы, открытые Фарадеем в 1833 г., строго выполняются для проводников второго рода. Наблюдаемые отклонения от законов Фарадея являются кажущимися. Они часто связаны с наличием неучтённых параллельных электрохимических реакций. Отклонения от закона Фарадея в промышленных установках связаны с утечками тока, потерями вещества при разбрызгивании раствора и т.д. В технических установках отношение количества продукта, полученного при электролизе, к количеству, вычисленному на основе закона Фарадея, меньше единицы и называется выходом по току. При тщательных лабораторных измерениях для однозначно протекающих электрохимических реакций выход по току равен единице (в пределах ошибок опыта). Закон Фарадея точно соблюдается, поэтому он лежит в основе самого точного метода измерения количества электричества, прошедшего через цепь, по количеству выделенного на электроде вещества. Для таких измерений используют серебряный или медный, а также йодный и газовый кулонометры (кулонометрия). 3. Теория электролитической диссоциации Аррениуса, ее недостатки. Причины электролитической диссоциации. ТЕОРИЯ ЭЛЕКТРОЛИТИЧЕСКОЙ ДИССОЦИАЦИИ АРРЕНИУСА. Электролитами называются вещества, которые при растворении в какой-либо среде распадаются на ионы независимо от того пропускается ли через систему электрический ток или нет. К электролитам относятся кислоты, основания и большинство неорганических и органических солей. Самопроизвольный процесс распада электролита в растворах на ионы называется электролитической диссоциацией. Для электролитов понижение температуры замерзания и осмотическое давление значительно больше соответствующих величин для неэлектролитов. В уравнение для осмотического давления Вант-Гофф ввел эмпирический коэффициент i 1, физический смысл которого стал понятен с появлением теории электролитической диссоциации: = icRT Теория электролитической диссоциации была предложена Аррениусом (1884-1887), развившим отдельные высказывания ряда ученых. Основные положения теории Аррениуса: Соли, кислоты, основания при растворении в воде и некоторых других полярных растворителях частично или полностью распадаются (диссоциируют) на ионы. Эти ионы существуют в растворе независимо от того, проходит через раствор электрический ток или нет. Вследствие этого число независимо движущихся частиц растворенного вещества больше, чем при отсутствии диссоциации, а величины коллигативных свойств растворов возрастают прямо пропорционально числу частиц. Коллигативными называют свойства разбавленных растворов, которые при данных условиях оказываются равными и независимыми от химической природы растворённого вещества, а зависят только от количества его частиц в растворе и природы растворителя. Наряду с процессом диссоциации в растворе идет обратный процесс ассоциация ионов в молекулы. В качестве меры электролитической диссоциации Аррениус ввел величину степени диссоциации , определяемую как долю молекул, распавшихся на ионы: N' c' = = N c Для любой обратимой реакции электролитической диссоциации: К+А- +Кz+ + -Azсумма + + - равна общему числу ионов, образующихся при диссоциации одной молекулы, которое равно коэффициенту Вант-Гоффа i: i = 1 + (+ + - 1) = 1 + ( 1) Определив коэффициент i, можно по этому уравнению вычислить степень диссоциации , если известна величина . По мере увеличения разведения коэффициент Вант-Гоффа приближается к простому целому числу (2, 3, 4 в зависимости от числа ионов, образующихся из одной молекулы вещества). Диссоциация растворенных веществ на ионы подчиняется тем же законам химического равновесия, что и др. реакции: K A z Кд = K z A Где Кд константа диссоциации, выраженная через концентрации. Диссоциация сильных электролитов равна 100% или почти 100%, так что концентрации ионов можно считать равными молярности растворенного вещества, умноженной на z. При диссоциации слабого электролита устанавливается равновесие между недиссоциированными молекулами и ионами. Рассмотрим простейший пример, когда молекула распадается только на два иона: СН3СООН СН3СОО- + Н+ с с CH COO H CH COOH Кд = с 3 3 = с c c ; c c 2c Кс = 1 Последнее равенство является простейшей формой закона разведения Освальда (1888). Чем больше Кс, тем выше степень диссоциации. Т.о., величина Кс может служить мерой силы кислоты, т.е. мерой кислотности. Для электролитов средней силы (Н3РО4 первая ступень, Са(ОН)2, СНСl2СООН) значения Кс лежат в пределах от 10-2 до 10-4; для слабых электролитов (СН3СООН, NН4ОН) Кс = 10-5 10-9; при Кс 10-10 электролит считается очень слабым (Н2О, С6Н5ОН, С6Н5NН2, НСN). Если очень мала, то ее величиной можно пренебречь по сравнению с 1, и формула примет вид: Кс = 2с; = Kc c Если электролит распадается больше чем на два иона, то зависимость Кс от усложняется: СаCl2 Ca2+ + 2Clс (1- ) с 2с Ca Cl 2 2 Кс = CaCl 2 2 4 3c2 c 2 c = = c1 1 При малой : = 3 Kc 4 c2 Величина Кс является постоянной только для очень разбавленных растворов, коэффициенты активности которых можно считать равными 1. Вообще же Кс (как и ) зависит от концентрации раствора; Кс иногда еще называют классической константой диссоциации. Кс () зависит также от температуры: зависимость Кс слабых кислот в воде проходит через максимум. Это можно объяснить влиянием двух противоположно направленных воздействий. С одной ст., всякая диссоциация протекает с поглощением тепла, и, следовательно, при повышении Т равновесие должно смещаться в сторону большей диссоциации. С др. ст., при повышении Т диэлектрическая проницаемость воды, служащей растворителем, уменьшается, а это способствует воссоединению ионов. Кс максимальна при той Т, при которой влияние второго фактора начинает преобладать. Обычно изменение Кд с повышением Т невелико. НЕДОСТАТКИ ТЕОРИИ АРРЕНИУСА. В теории электролитов очень важным является вопрос о распределении ионов в растворе. По первоначальной теории электролитической диссоциации, основанной на физической теории растворов Вант-Гоффа, считалось, что ионы в растворах находятся в состоянии беспорядочного движения в состоянии, подобном газообразному. Однако представление о беспорядочном распределении ионов в растворе не соответствует действительности, так как онo не учитывают электростатического взаимодействия между ионами. Электрические силы проявляются на относительно больших расстояниях, и в сильных электролитах, где диссоциация велика, а концентрация ионов значительна и расстояния между ними невелики, электростатическое взаимодействие между ионами настолько сильно, что оно не может не сказываться на характере их распределения. Возникает тенденция к упорядоченному распределению, аналогичному распределению ионов в ионных кристаллах, где каждый ион окружён ионами противоположного знака. Распределение ионов будет определяться соотношением электростатической энергии и энергии хаотического движения ионов. Эти энергии сравнимы по величине, поэтому реальное распределение ионов в электролите является промежуточным между беспорядочным и упорядоченным. В этом заключается своеобразие электролитов и трудности, возникающие при создании теории электролитов. Около каждого иона образуется своеобразная ионная атмосфера, в которой преобладают ионы противоположного (по сравнению с центральным ионом) знака. Теория Аррениуса не учитывала этого обстоятельства, и многие выводы этой теории оказались в противоречии с опытом. В качестве одной из количественных характеристик электролита теория Аррениуса предлагает степень электролитической диссоциации , определяющую долю ионизированных молекул в данном растворе. В согласии с ее физическим смыслом не может быть больше 1 или меньше 0; при заданных условиях она должна быть одной и той же, независимо от метода ее измерения (по измерению электропроводности, осмотического давления или ЭДС). Однако на практике значения , полученные разными методами, совпадают только для разбавленных растворов слабых электролитов; для сильных электролитов расхождение тем больше, чем больше концентрация электролита, причем в области высоких концентраций становится больше 1. Следовательно, не может иметь того физического смысла, который ей приписывался теорией Аррениуса. Второй количественной характеристикой по теории Аррениуса является константа диссоциации; она должна быть постоянной для данного электролита при заданных Т и Р, независимо от концентрации раствора. На практике только для разбавленных растворов очень слабых электролитов Кдис остается при разбавлении более или менее постоянной. Т.о., теория электролитической диссоциации приложима только к разбавленным растворам слабых электролитов. Рассмотрим процесс диссоциации одно-однозарядного электролита: KtAn = Kt+ + An-. (1) Если концентрация электролита в растворе С и степень его диссоциации α, количество распавшегося на ионы электролита составит Сα. Тогда концентрация катионов и анионов в растворе составит Сα, а концентрация непродиссоциирующего элекролита составит С – Сα. Уравнение закона действующих масс для реакции (1) выражается уравнением: С Kt C An C C К = C KtAn = C C , (2) где К – константа равновесия или константа диссоциации электролита. После преобразования получим: С 2 K = 1 . (3) Поскольку константа равновесия К зависит только от температуры, то можно заключить, что при разведении (уменьшении концентрации) электролита степень его диссоциации увеличивается. Разведением называют объём электролита, в котором содержится один граммэквивалент электролита, т.е. 1 V = С , (4) где V – разведение, л/г-экв. Тогда уравнение (3) можно представить в виде: 2 К = V (1 ) . (5) Уравнения (3) и (5) часто называют законом разведения Оствальда. В результате процесса диссоциации в растворе электролитов увеличивается число частиц. Это увеличение характеризует изотонический коэффициент Вант – Гоффа. Общее число частиц в единице объёма раствора для электролита с концентрацией С и степенью диссоциации α, распадающегося при диссоциации на k ионов, составит: (С – Сα) + Сkα = С [1 – α(k – 1)]= iC, (6) где i = 1 – α(k – 1) – изотонический коэффициент, который всегда больше единицы(i > 1). Тогда для степени диссоциации электролита можно записать: i 1 α = k 1 . (7) Поскольку степень диссоциации слабых электролитов мала, то справедливо отношение α 1, тогда уравнение (3) можно представить в виде: K = Cα2, (8) откуда получим: К α = С . (9) Классическая теория электролитической диссоциации позволила объяснить ряд опытных данных. Так тепловой эффект реакций нейтрализации сильных кислот и оснований для всех кислот и оснований, отнесенный к одному грамм – молю воды, одинаков и равен тепловому эффекту реакции образования воды из ионов: Н+ + ОН- = Н 2О, (10) что указывает на ионный характер реакции нейтрализации. Электропроводность растворов электролитов увеличивается с разбавлением раствора. электролит активность Это связано с тем, что при разбавлении увеличивается степень диссоциации электролита, что, в свою очередь, приводит к увеличению числа ионов в растворе вследствие чего и возрастает электропроводность раствора. ПРИЧИНЫ ЭЛЕКТРОЛИТИЧЕСКОЙ ДИССОЦИАЦИИ. Твердые вещества, при растворении которых в воде и других полярных растворителях образуются электролиты, являются, как правило, кристаллическими телами, имеющими ионные или близкие к ионным решётки. В чисто ионных решётках не существует молекул вещества. Ионы противоположных знаков, составляющие такую решётку, связаны между собой большими электростатическими силами. При переходе ионов в раствор энергии электростатического взаимодействия ионов в решётке противопоставляется энергия взаимодействия ионов с дипольными молекулами растворителя, который втягивает ионы решётки в раствор. При этом ионы окружаются молекулами растворителя, образующими вокруг иона сольватную (в частном случае гидратную) оболочку. Энергия взаимодействия ионов различных знаков, перешедших в раствор и окружённых сольватными оболочками, уменьшается по сравнению с энергией их взаимодействия в решётке обратно пропорционально диэлектрической проницаемости растворителя D в соответствии с законом Кулона: z1z2 e2 E= Dr где z1 и z2 число элементарных зарядов катиона и аниона; r расстояние между катионом и анионом. Взаимодействие электрически заряженных тел в воздухе практически не отличается от их взаимодействия в вакууме. Если заряженные тела находятся в воде, керосине, масле или какойнибудь другой непроводящей среде, то модуль сил их взаимодействия оказывается меньше, чем в вакууме. Чтобы учесть влияние среды, ввели её специальную характеристику, называемую диэлектрической проницаемостью. Диэлектрическая проницаемость вещества — физическая величина, показывающая, во сколько раз модуль сил электростатического взаимодействия зарядов в данной однородной среде меньше модуля сил взаимодействия этих же зарядов в вакууме: , где F0 и F — модули сил электростатического взаимодействия зарядов в вакууме и в однородной среде соответственно. С учётом соотношения закон Кулона можно записать следующим образом: Диэлектрическая проницаемость вакуума равна 1. За 1 принимают и диэлектрическую проницаемость воздуха, поскольку её значение (при нормальном атмосферном давлении) 1,0006. Диэлектрические проницаемости других однородных сред всегда больше единицы. Например, у воды диэлектрическая проницаемость 81, у глицерина — 56, а у керосина — 2. Если энергия взаимодействия ионов с растворителем становится соизмеримой с энергией ионов в кристаллической решётке, то происходит растворение с диссоциацией. Надо помнить при этом, что при растворении должен уменьшиться изобарный потенциал G системы, а внутренняя энергия (и энтальпия) может как уменьшаться, так и увеличиваться (отрицательная и положительная теплоты растворения). Взаимодействие дипольных молекул растворителя с элементами кристаллической решётки может привести к образованию электролита даже при растворении веществ, имеющих молекулярную решётку, решётку промежуточного типа или находящихся в газообразном состоянии (атомы в молекулах газа связаны ковалентно). Ясно, что для осуществления электролитической диссоциации определяющую роль играет взаимодействие ионов с растворителем (в водных растворах гидратация, в общем случае сольватация). На важное значение гидратации ионов впервые указали И.А. Каблуков и В.А. Кистяковский. Они положили начало развитию теории электролитов в направлении, которое указывал Д.И. Менделеев, то есть объединили так называемую сольватную теорию и физическую теорию Вант-Гоффа Аррениуса. Связь между диэлектрической проницаемостью D растворителя и его способностью образовывать растворы, проводящие электрический ток, отмечалась давно. Вода, диэлектрическая проницаемость которой D = 81 при 18C, а также HCN (D=107 при 25C) и HCOOH (D=57 при 25С) принадлежат к растворителям, вызывающим сильную диссоциацию. Низшие спирты и кетоны, уксусная кислота, пиридин имеют диэлектрические проницаемости в пределах 2035 и также способны образовывать электролиты, хотя и в меньшей степени, чем вода. Кроме величины диэлектрической проницаемости важное значение имеет взаимодействие молекул растворителя с молекулами растворённого вещества. Это взаимодействие нередко приводит к образованию новых молекул или молекулярных комплексов, которые в данном растворителе способны диссоциировать на ионы. 4. Удельная электропроводность. Ее зависимость от концентрации и температуры. Кривая зависимости удельной электропроводности растворов от концентрации обычно имеет максимум (четко выраженный для сильных электролитов и сглаженный для слабых). Наличие максимумов на кривых с можно объяснить следующим образом. В разбавленных растворах сильных электролитов ( = 1) электропроводность растет пропорционально числу ионов, которое, в свою очередь, растет с концентрацией. В концентрированных растворах сильных электролитов ионная атмосфера существенно уменьшает скорость движения ионов, и электропроводность падает. В слабых электролитах плотность ионной атмосферы мала и скорость движения ионов мало зависит от концентрации, однако с увеличением концентрации раствора заметно уменьшается степень диссоциации, что приводит к уменьшению концентрации ионов и падению электропроводности. сильный эл-т слабый эл-т С Зависимость удельной электропроводности от концентрации электролита 5. Эквивалентная электропроводность. Ее зависимость от концентрации и температуры. Подвижность ионов. Аномальная подвижность ионов гидроксония и гидроксида. сильный эл-т слабый эл-т С Зависимость эквивалентной электропроводности от концентрации электролита Эквивалентная электропроводность в см2/(г-эквОм) это электропроводность такого объема ( см3) раствора, в котором содержится 1 г-экв растворенного вещества, причем электроды находятся на расстоянии 1 см друг от друга. Найдем связь между и . Представим себе погруженные в раствор параллельные электроды на расстоянии 1 см, имеющие весьма большую площадь. Электропроводность раствора, заключенного между поверхностями таких электродов, имеющими площадь, равную см2, и есть эквивалентная электропроводность раствора. Объем раствора между этими площадями электродов равен см3 и содержит 1 г-экв соли. Величина , равная 1000/с см3/г-экв, называется разведением. Таким образом: = ; = 1000 c Мольная электропроводность электролита это произведение эквивалентной электропроводности на число грамм-эквивалентов в 1 моль диссоциирующего вещества. Зависимость эквивалентной электропроводности от концентрации: 1. Зависимость с: с увеличением с величина уменьшается сначала резко, а затем более плавно. 2. Зависимость c : для сильных электролитов соблюдается медленное линейное уменьшение с увеличением c , что соответствует эмпирической формуле Кольрауша: = А c предельная эквивалентная электропроводность при бесконечном разведении: с 0, . 3. Зависимость : значение сильных электролитов растет с увеличением и асимптотически приближается к . Для слабых электролитов значение также растет с увеличением , но приближение к пределу и величину предела в большинстве случаев практически нельзя установить. сильный эл-т сильный эл-т слабый эл-т слабый эл-т C Все вышесказанное касалось электропроводности водных растворов. Для электролитов с другими растворителями рассмотренные закономерности сохраняются, но имеются и отступления от них, например, на кривых с часто наблюдается минимум (аномальная электропроводность). Примечания: Эквивалентная концентрация (г-экв/см3); Напряженность поля, В/см; Абсолютные подвижности ионов, см2/(с*В) - скорости ионов в стандартных условиях; Подвижность является важнейшей характеристикой ионов, отражающей их специфическое участие в электропроводности электролита. В водных растворах все ионы, за исключением ионов Н3О+ и ОН-, обладают подвижностями одного порядка; их абсолютные подвижности (u и v) равны нескольким см в час. Эквивалентная электропроводность растворов солей выражается величинами порядка 100 - 130 см2/(г-эквОм). Ввиду исключительной подвижности иона гидроксония величины для кислот в 3-4 раза больше, чем для солей; щелочи занимают промежуточное положение. Движение иона можно уподобить движению макроскопического шарика в вязкой среде и применить в этом случае формулу Стокса: u= ze E 6r l где е заряд электрона; z число элементарных зарядов иона; r эффективный радиус иона; коэффициент вязкости; E/l напряженность поля. Движущую силу напряженность поля Е/l при вычислении абсолютных подвижностей принимаем равной единице. Следовательно, скорость движения ионов обратно пропорциональна их радиусу. Рассмотрим ряд Li+, Na+, K+. Так как в указанном ряду истинные радиусы ионов увеличиваются, то подвижности должны уменьшаться в той же последовательности. Однако в действительности это не так. Подвижности увеличиваются при переходе от Li+ к K+ почти в два раза. Из этого можно сделать заключение, что в растворе и ионной решётке ионы обладают разными радиусами. При этом чем меньше истинный (кристаллохимический) радиус иона, тем больше его эффективный радиус в электролите. Это явление можно объяснить тем, что в растворе ионы не свободны, а гидратированы. Тогда эффективный радиус движущегося в электрическом поле иона будет определяться в основном степенью его гидратации, то есть количеством связанных с ионом молекул воды. Связь иона с молекулами растворителя ионно-дипольная, а так как напряжённость поля на поверхности иона лития гораздо больше, чем на поверхности иона калия, то степень гидратации иона лития больше степени гидратации иона калия. Согласно формуле Стокса, многозарядные ионы должны обладать большей подвижностью, чем однозарядные. Однако скорости движения многозарядных ионов мало отличаются от скоростей движения однозарядных, что, очевидно, объясняется большей степенью их гидратации. ПОДВИЖНОСТЬ ИОНОВ ГИДРОКСОНИЯ И ГИДРОКСИЛА. Аномально высокая подвижность ионов гидроксония и гидроксила была отмечена давно. Раньше считали, что в растворе существуют ионы водорода, большая скорость движения которых объясняется исключительно малым радиусом ионов. Установили, что в растворе имеются не ионы водорода H+, а ионы гидроксония H3O+. Эти ионы, так же, как и ионы гидроксила, гидратированы, и эффективные радиусы их имеют тот же порядок, что и радиусы других ионов. Следовательно, если бы механизм переноса электричества этими ионами был обычным, то подвижность их не отличалась бы существенно от подвижностей других ионов. Это и наблюдается в действительности в большинстве неводных растворов. Аномально высокая подвижность H3O+ и OH- проявляется только в растворах в воде и простейших спиртах, что, очевидно, связано с тем, что они являются ионами самого растворителя воды. Известно, что процесс диссоциации воды протекает по схеме H2O + H2O = OH- + H3O+ H+ и сводится к переходу протона от одной молекулы воды к другой. Образовавшиеся ионы гидроксония непрерывно обмениваются протонами с окружающими молекулами воды, причём обмен протонами происходит хаотически. Однако при создании разности потенциалов кроме беспорядочного движения возникает и направленное: часть протонов начинает двигаться к катоду и, следовательно, переносит электричество. Таким образом, электричество переносится в основном не ионами гидроксония, а протонами, перескакивающими от одной молекулы воды к другой ориентированно. Благодаря описанному движению протонов увеличивается электропроводность раствора, потому что протоны имеют очень малый радиус и проходят не весь путь до катода, а лишь расстояния между молекулами воды. Этот тип проводимости можно назвать эстафетным или цепным. Аналогично можно объяснить большую подвижность гидроксильных ионов, только в этом случае переход протонов происходит не от ионов гидроксония к молекулам воды, а от молекул воды к ионам гидроксила, что приводит к кажущемуся перемещению ионов гидроксила по направлению к аноду. Ионы гидроксила действительно появляются в анодном пространстве, но это объясняется в основном не движением их, а перескоком протонов по направлению к катоду. Конечно, ионы H3O+ и OH-, как таковые, также движутся при создании разности потенциалов между электродами и переносят электричество, но вклад их в электропроводность приблизительно такой же, как и вклад других ионов. Большая электропроводность кислот и оснований объясняется именно цепным механизмом электропроводности с участием протонов. 6. Связь между подвижностью ионов и их концентрацией. Электрофоретический и релаксационный эффекты. СВЯЗЬ МЕЖДУ ПОДВИЖНОСТЬЮ ИОНОВ И ИХ КОНЦЕНТРАЦИЕЙ. Теория электролитической диссоциации Аррениуса не учитывала влияния концентрации на подвижность ионов, хотя, как выяснилось, влияние концентрации на подвижность может быть весьма существенным. Кольрауш вывел эмпирическое уравнение, связывающее эквивалентную электропроводность сильных электролитов с концентрацией: = А c о о Так как = + + - и = + + -, то, следовательно, + = о+ В1 c и - = о В2 c , где В1 + В2 = А. Дебай и Гюккель объясняли уменьшение подвижности ионов и эквивалентной электропроводности сильных электролитов с увеличением концентрации наличием ионной атмосферы. Действительно, каждый ион окружён ионной атмосферой, состоящей преимущественно из ионов противоположного знака, плотность которой увеличивается с повышением концентрации электролита. При наложении электрического поля ион начинает двигаться в одну сторону, а ионная атмосфера в противоположную. Движение ионов разных зарядов в противоположных направлениях создаёт как бы дополнительное трение, которое и уменьшает абсолютную скорость движения ионов. Этот эффект торможения носит название электрофоретического эффекта. По мере увеличения концентрации плотность ионной атмосферы увеличивается, следовательно, увеличивается и тормозящий электрофоретический эффект. При движении ион покидает свою ионную атмосферу и непрерывно на пути своего движения создаёт новую. Этот процесс разрушения старой и образования новой ионной атмосферы протекает хотя и быстро, но не мгновенно, вследствие чего при движении иона нарушается симметричность ионной атмосферы, причём плотность её больше позади движущегося иона. Появление асимметрии ионной атмосферы также вызывает некоторое торможение движения иона, которое получило название эффекта асимметрии или релаксации. Таким образом, из-за наличия ионной атмосферы при движении иона возникают два тормозящих эффекта: электрофоретический, обусловленный движением ионной атмосферы в сторону, противоположную направлению движения иона, и эффект релаксации, обусловленный асимметрией ионной атмосферы. 1) С ростом концентрации ионов λi и их подвижность уменьшается, что обусловлено наличием ионной атмосферы, плотность которой возрастает с увеличением концентрации; 2) Из-за наличия ионной атмосферы возникают два тормозящих эффекта – электрофоретический и релаксационный. Электрофоретический эффект – обусловлен движением ионной атмосферы в сторону противоположную от центра иона. Это создает дополнительное “трение” и замедляет скорость движения ионов. Релаксационный эффект – обусловлен тем, что при движении ион смещается из центра своей ионной атмосферы, в результате чего нарушается ее симметрия, и плотность заряда позади иона оказывается больше, чем впереди его. В результате на центральный ион действует возвращающая сила, замедляющая его движение. 7. Эффекты Вина и Дебая-Фалькенгагена. Уравнение Онзагера. Убедительным подтверждением правильности представлений Дебая и Гюккеля является так называемый эффект Вина. Если уменьшение подвижности ионов с увеличением концентрации объясняется наличием ионной атмосферы, то уничтожение последней должно привести к возрастанию подвижности и электропроводности до предельного значения. Поскольку скорость движения иона пропорциональна напряжению, а скорость образования ионной атмосферы является конечной величиной, то путём увеличения напряжённости можно добиться такой большой скорости движения ионов, при которой ионная атмосфера уже не будет успевать образовываться. Тогда ионы будут обладать максимальной скоростью движения и предельной подвижностью. Это и было установлено Вином, который, увеличив напряжённость поля до 200000 в/см, наблюдал увеличение эквивалентной электропроводности до предельного значения . В 1928 г. Дебай и Фалькенгаген теоретически рассмотрели влияние частоты переменного тока на электропроводность электролитов и установили, что при увеличении частоты выше некоторого значения должно наблюдаться заметное возрастание электропроводности. Явление увеличения электропроводности с частотой получило название частотного эффекта или дисперсии электропроводности. При достаточно большой частоте переменного тока взаимные смещения иона и ионной атмосферы настолько малы, что ионная атмосфера практически симметрична, а потому тормозящий эффект релаксации должен исчезнуть. При больших частотах эффект релаксации исчезает. Электрофоретический эффект остаётся, так как ионная атмосфера не уничтожается. Следовательно, частотный эффект должен быть меньшим, чем эффект Вина. Сопоставляя значения того и другого, можно расчленить суммарный эффект уменьшения электропроводности на составляющие. Эффект Вина возникает при полном уничтожении ионной атмосферы, а следовательно, и обоих эффектов торможения. Частотный эффект объясняется лишь исчезновением симметрии ионной атмосферы. Опыт показывает, что последний эффект примерно в 3 раза слабее, чем эффект Вина, то есть электрофоретический эффект в 2 раза сильнее эффекта релаксации. Опыты Вина и Дебая-Фалькенгагена являются убедительным экспериментальным доказательством реального существования ионной атмосферы и позволяют представить себе характер её строения. Представление о ионной атмосфере является одним из фундаментальных положений электростатической теории электролитов. В дальнейшем Онзагер вывел теоретическое уравнение, которое количественно связывает эквивалентную электропроводность с концентрацией и позволяет вычислить электрофоретический и релаксационный эффекты. Для бинарных одновалентных водных электролитов уравнение Онзагера имеет вид: 8,2 105 82,4 c = 3/ 2 DT 1/ 2 DT где первое слагаемое в квадратных скобках характеризует эффект релаксации, второе слагаемое характеризует электрофоретический эффект; D диэлектрическая проницаемость, коэффициент вязкости, Т температура, с концентрация. Теоретическое уравнение Онзагера согласуется с эмпирической формулой Кольрауша ( = А c ) в интервале средних концентраций. ЗАВИСИМОСТЬ ПОДВИЖНОСТИ ИОНОВ ОТ ТЕМПЕРАТУРЫ. Предельная подвижность ионов, а также удельная электропроводность электролитов всегда увеличиваются с повышением температуры (в противоположность электропроводности металлов, которая уменьшается с повышением температуры). Температурный коэффициент подвижности оказывается довольно большим; при нагревании раствора на 1oС подвижность, а следовательно, и электропроводность возрастают примерно на 2 %. Наибольший температурный коэффициент характерен для ионов с относительно малой подвижностью и наоборот. Наличие положительного температурного коэффициента подвижности ионов, по-видимому, объясняется уменьшением вязкости с температурой. Так как = о+ + о-, то эквивалентная электропроводность при бесконечном разведении с температурой всегда возрастает. При конечной концентрации связь с подвижностью несколько сложнее. Для слабого электролита = (+ + -). Если с повышением температуры подвижности ионов возрастают, то степень диссоциации может и уменьшаться, поскольку диэлектрическая проницаемость раствора при нагревании уменьшается, то есть силы взаимодействия между ионами увеличиваются. Следовательно, кривая зависимости электропроводности от температуры может иметь максимум. Аналогичное явление наблюдается и в сильных электролитах. 8. Числа переноса, методы определения чисел переноса. Закон Кольрауша Одним из важнейших понятий в электрохимии является число переноса ионов. В электролитах электричество переносится одновременно положительными и отрицательными ионами, поэтому возникает вопрос, каково участие в этом процессе ионов каждого знака. Количество переносимого электричества определяется концентрацией ионов и скоростью их движения; когда концентрации катионов и анионов одинаковы, участие их в переносе электричества зависит лишь от относительной скорости их движения. Т.к. скорости движения катионов и анионов могут быть существенно различными, потому и числа переноса должны быть разными. Это было установлено Гитторфом (1854). Числом переноса ионов называется доля прошедшего через электролит электричества, перенесенная данным родом ионов: I I I I t+ = , t- = , t+ + t- = = 1 I I I u v qcFE uqc FE vqc FE I+ = , I- = , I = I+ + I- = l t+ = l I u = = I uv l , t- = I v = = I uv Таким образом, число переноса равно отношению скорости движения (или подвижности) данного иона к сумме скоростей движения (или подвижностей) катиона и аниона. Т.к. подвижности катиона и аниона изменяются с концентрацией и температурой в общем случае неодинаково, то и числа переноса зависят от концентрации и температуры, хотя эта зависимость более слабая. Выведенное соотношение позволяет вычислить числа переноса, если известны значения подвижностей ионов. С др. стороны, опытное определение чисел переноса дает возможность вычислить подвижности. Видно, что число переноса не является характеристикой только данного иона, т.к. зависит от подвижности парного с ним иона. Экспериментально числа переноса определяются по изменению концентрации ионов у электродов (метод Гитторфа). Рассмотрим схему движения ионов (переноса электричества) в растворе HCl при электролизе. Разделим мысленно ванну с электролитом на три отделения: I анодная часть (анолит), II центральная часть, срединное пространство, III катодная часть (католит). В процессе электролиза в отделении II концентрация электролита не изменяется, в отделениях I и III изменяется. Схема А: до электролиза концентрация раствора во всех отделениях одинакова (по 6 пар ионов). Схема Б : абсолютная скорость движения Н+ приблизительно в 5 раз больше, чем скорость Cl-, поэтому в течение определенного промежутка времени при электролизе ионы Н+ пройдут слева направо путь, в 5 раз больший, чем ионы Сl- за то же время справа налево. В результате в катодном пространстве появится 6 лишних ионов водорода, а в анодном столько же лишних ионов хлора (в катодном пространстве появятся 3 лишних пар ионов водорода, а в анодном 3 лишних пар ионов хлора). Схема В: эти ионы разряжаются на электродах и выделяются в виде газов. В анодном пространстве остается одна пара ионов, в катодном 5 пар ионов. Убыль электролита у анода са (5 пар ионов) в 5 раз больше убыли электролита у катода ск (1 пара ионов). Отношение са/ск равно отношению абсолютных скоростей катиона и аниона и равно отношению их подвижностей. Числа переноса определяются из соотношений: ca ck t+ = = , t- = = ca ck ca ck и в рассмотренном примере t+(H+) 5/6 = 0.83, t-(Cl-) 1/6 = 0.17, откуда видно, что количество ионов данного типа, участвующих в переносе электричества, никак не связано с количеством ионов, передавших свой заряд электроду. Определенные по методу Гитторфа числа переноса называются кажущимися числами переноса; они не являются истинными, т.к. этот метод не учитывает сольватации ионов. Измеряемые в методе Гитторфа концентрации определяются не только количеством катионов и анионов, но и количеством растворителя, перенесенного этими ионами в виде сольватных оболочек. Оболочки ионов разных знаков неодинаковы по величине. Существование рассмотренного эффекта можно легко установить, прибавив к электролиту недиссоциирующее на ионы вещество, например, сахар или мочевину. Учитывая изменение концентрации прибавленного неэлектролита при определении чисел переноса, можно ввести поправку на перенос воды из анодного пространства в катодное в виде сольватных оболочек и найти истинные числа переноса + и -. Но обычно в значения чисел переноса, найденные опытным путем по методу Гитторфа, поправки не вводятся. Зависимость чисел переноса от концентрации обычно невелика. Однако в некоторых случаях число переноса сильно изменяется с концентрацией и может оказаться равным нулю и даже меньше нуля (например, для концентрированного раствора CdI2 t+ 0). Это можно объяснить образованием комплексных анионов (CdI42-). Влияние температуры на числа переноса незначительно. Во многих случаях числа переноса при повышении температуры приближаются к 0,5, т.е. подвижности катиона и аниона становятся почти одинаковы. Растворы одной и той же соли в разных растворителях имеют различные числа переноса; это объясняется, в основном, различной степенью сольватации катионов и анионов в зависимости от растворителя. 9. Метод активности в термодинамике растворов электролитов, средний коэффициент активности электролита. Теория Дебая-Хюккеля, допущения и три приближения. Активность растворенной соли а может быть определена по давлению пара, температуре затвердевания, по данным о растворимости, методом ЭДС. Все методы определения активности соли приводят к величине, характеризующей реальные термодинамические свойства растворенной соли в целом, независимо от того, диссоциирована она или нет. Однако в общем случае свойства различных ионов неодинаковы, и можно ввести и рассматривать термодинамические функции отдельно для ионов разных видов: + = +о + RT ln a+ = +o + RT ln m+ + RT ln + - = -о + RT ln a- = -o + RT ln m- + RT ln - где + и - практические коэффициенты активности (коэффициенты активности при концентрациях, равных моляльности m). Но термодинамические свойства различных ионов не могут быть определены порознь из опытных данных без дополнительных допущений; мы можем измерить только средние термодинамические величины для ионов, на которые распадается молекула этого вещества. Пусть диссоциация соли происходит по уравнению: АВ = Аz + Bz При полной диссоциации m = m , m = m. Пользуясь уравнениями ГиббсаДюгема, можно показать : аа а = const Стандартные состояния для нахождения величин активностей определяются так: lim a+ m+ = m при m 0 , lim a- m- = - m при m 0 Стандартное состояние для а выбирается так, чтобы const была равна 1. Тогда: аа = а Т.к. нет методов экспериментального определения значений а+ и а- в отдельности, то вводят среднюю ионную активность а, определяемую соотношением: а = а Т.о., мы имеем две величины, характеризующие активность растворенной соли. Первая из них это мольная активность, т.е. активность соли, определяемая независимо от диссоциации; она находится теми же экспериментальными методами и по тем же формулам, что и активность компонентов в неэлектролитах. Вторая величина средняя ионная активность а . Введем теперь коэффициенты активности ионов + и -, среднюю ионную моляльность m и средний ионный коэффициент активности : a+ = +m+, a- = -m-, m = (m+m)1/ = ()1/ m = ()1/ Очевидно: a = ()1/ ()1/ m = m Т.о., основные величины связаны соотношениями: a = m = ()1/ m = L m где L = ()1/ и для солей каждого определенного типа валентности является величиной постоянной. Величина является важной характеристикой отклонения раствора соли от идеального состояния. В растворах-электролитах, как и в растворах-неэлектролитах, могут быть использованы следующие активности и коэффициенты активности: a N рациональный коэффициент активности (практически не применяется); N a m = практический коэффициент активности (средний моляльный); m a c f = средний мольный коэффициент активности. c = Основными методами измерения величины являются криоскопический и метод ЭДС. Многочисленные исследования показали, что кривая зависимости от концентрации раствора (m) имеет минимум. Если изображать зависимость в координатах lg m , то для разбавленных растворов зависимость оказывается линейной. Наклон прямых, соответствующих предельному разбавлению, одинаков для солей одного валентного типа. Присутствие в растворе других солей изменяет коэффициент активности данной соли. Суммарное влияние смеси солей в растворе на коэффициент активности каждой из них охватывается общей закономерностью, если суммарную концентрацию всех солей в растворе выразить через ионную силу. Ионной силой I (или ионной крепостью) раствора называется полусумма произведений концентрации каждого иона на квадрат числа его заряда (валентности), взятая для всех ионов данного раствора. Если использовать моляльность как меру концентрации, то ионная сила раствора определяется выражением: 1 2 I = mi zi 2 i где i индексы ионов всех солей в растворе; mi = i m. Льюис и Рендалл открыли эмпирический закон ионной силы: средний ионный коэффициент активности диссоциирующего на ионы вещества является универсальной функцией ионной силы раствора, т.е. в растворе с данной ионной силой все диссоциирующие на ионы вещества имеют коэффициенты активности, не зависящие от природы и концентрации данного вещества, но зависящие от числа и валентности его ионов. Закон ионной силы отражает суммарное взаимодействие ионов раствора с учетом их валентности. Этот закон точен лишь при очень малых концентрациях (m = <0,02); уже при умеренных концентрациях он верен лишь приблизительно. В разбавленных растворах сильных электролитов: lg = А I ТЕОРИЯ ЭЛЕКТРОЛИТОВ ДЕБАЯ И ГЮККЕЛЯ. Основные положения современной теории растворов электролитов были сформулированы в 1923 г. Дебаем и Гюккелем. Для статистической теории электролитов исходным является следующее положение: ионы распределены в объеме раствора не хаотически, а в соответствии с законом кулоновского взаимодействия. Вокруг каждого отдельного иона существует ионная атмосфера (ионное облако) сфера, состоящая из ионов противоположного знака. Ионы, входящие в состав сферы, непрерывно обмениваются местами с другими ионами. Все ионы раствора равноценны, каждый из них окружен ионной атмосферой, и в то же время каждый центральный ион входит в состав ионной атмосферы какоголибо другого иона. Существование ионных атмосфер и есть тот характерный признак, который, по Дебаю и Гюккелю, отличает реальные растворы электролитов от идеальных. С помощью уравнений электростатики можно вывести формулу для электрического потенциала ионной атмосферы, из которой вытекают уравнения для средних коэффициентов активности в электролитах: e zi r e 1 = Dr D диэлектрическая проницаемость раствора; е заряд электрона; zi заряд иона; r координата (радиус). 4e 2 N A zi2 ni = DkT i 1/ 2 величина, зависящая от концентрации раствора, D и Т, но не зависящая от потенциала; имеет размерность обратной длины; характеризует изменение плотности ионной атмосферы вокруг центрального иона с увеличением расстояния r от этого иона. Величина 1/ называется характеристической длиной; ее можно отождествить с радиусом ионной атмосферы. Она имеет большое значение в теории растворов электролитов. Для коэффициента активности получено следующее выражение: lg f = Azz I (1) Коэффициент A зависит от Т и D: обратно пропорционален (DT)3/2. Для водных растворов 1-1 зарядных электролитов при 298 К, допуская равенство диэлектрических проницаемостей раствора и растворителя (78,54), можно записать: lg f = A I = A c = 0,51 c Т.о., теория Дебая и Гюккеля позволяет получить такое же уравнение для коэффициента активности, какое было эмпирически найдено для разбавленных растворов электролитов. Теория, следовательно, находится в качественном согласии с опытом. При разработке этой теории были сделаны следующие допущения: 1. Число ионов в электролите можно определить из аналитической концентрации электролита, т.к. он считается полностью диссоциированным ( 1). Теорию Дебая и Гюккеля поэтому иногда называют теорией полной диссоциации. Однако ее можно применить и в тех случаях, когда 1. 2. Распределение ионов вокруг любого центрального иона подчиняется классической статистике Максвелла-Больцмана. 3. Собственными размерами ионов можно пренебречь по сравнению с расстояниями между ними и с общим объемом раствора. Т.о., ионы отождествляются с материальными точками, и все их свойства сводятся лишь к величине заряда. Это допущение справедливо только для разбавленных растворов. 4. Взаимодействие между ионами исчерпывается кулоновскими силами. Наложение сил теплового движения приводит к такому распределению ионов в растворе, для которого характерна статистическая шаровая ионная атмосфера. Это допущение справедливо лишь для разбавленных растворов. При повышении концентрации среднее расстояние между ионами уменьшается, и наряду с электростатическими силами появляются другие силы, действующие на более близком расстоянии, в первую очередь силы Ван-дер-Ваальса. Возникает необходимость учета взаимодействия не только между данным ионом и его окружением, но и между любыми двумя соседними ионами. 5. При расчетах принимается, что диэлектрические проницаемости раствора и чистого растворителя равны; это справедливо только в случае разбавленных растворов. Т.о., все допущения Дебая и Гюккеля приводят к тому, что их теория может быть применима только к разбавленным растворам электролитов с ионами низкой валентности. Уравнение (1) соответствует этому предельному случаю и выражает так называемый предельный закон Дебая и Гюккеля или первое приближение теории Дебая и Гюккеля. Предельный закон Дебая-Гюккеля дает верные значения коэффициентов активности 1-1 зарядного электролита, особенно в очень разбавленных растворах. Сходимость теории с опытом ухудшается по мере увеличения концентрации электролита, увеличения зарядов ионов и уменьшения диэлектрической проницаемости растворителя, т.е. с ростом сил взаимодействия между ионами. Первая попытка усовершенствовать теорию Дебая и Гюккеля и расширить область ее применения была сделана самими авторами. Во втором приближении они отказались от представления об ионах как о материальных точках (допущение 3) и попытались учесть конечные размеры ионов, наделив каждый электролит некоторым средним диаметром а (при этом изменяется и допущение 4). Приписав ионам определенные размеры, Дебай и Гюккель учли тем самым силы некулоновского происхождения, препятствующие сближению ионов на расстояние, меньшее некоторой величины. Во втором приближении средний коэффициент активности описывается уравнением: lg f = Az z I (2) 1 Ba I где А сохраняет прежнее значение; а условно названо средним эффективным диаметром ионов, имеет размерность длины, фактически эмпирическая постоянная; В = / I , В слегка изменяется с Т. Для водных растворов произведение Ва близко к 1. Сохранив основные положения второго приближения теории, Гюккель учел уменьшение диэлектрической проницаемости с ростом концентрации растворов. Ее уменьшение вызывается ориентацией диполей растворителя вокруг иона, в результате чего снижается их реакция на эффект внешнего поля. Уравнение Гюккеля выглядит следующим образом: lg f = Az z I + CI (3) 1 Ba I где С эмпирическая константа. При удачном подборе значений В и С формула Гюккеля хорошо согласуется с опытом и широко используется при расчетах. При последовательном уменьшении ионной силы уравнение (3) последовательно переходит в формулу второго приближения теории Дебая и Гюккеля (уравнение (2)), а затем в предельный закон Дебая-Гюккеля (уравнение (1)). В процессе развития теории Дебая-Гюккеля и последовательного отказа от принятых допущений улучшается сходимость с опытом и расширяется область ее применимости, однако это достигается ценой превращения теоретических уравнений в полуэмпирические. 10. Ионное равновесие в растворах электролитов. Диссоциация воды. pH растворов. Буферные растворы. В основе ионных равновесий в растворах электролитов лежат положения классической теории диссоциации электролитов. Согласно этой теории кислотой называется вещество, которое при диссоциации распадается на ионы водорода и кислотного остатка: HAn = H+ + An-, а основанием – вещество, которое при диссоциации распадается на ионы металла и гидроксила: MeOH = Me+ + OH-. К процессу диссоциации применим закон действующих масс: С Н С An С Ме С ОН Kк = C HAn , Кос. = С МеОН . Если концентрация кислоты или основания С, а степень диссоциации α, то уравнения для константы диссоциации кислоты или основания примут вид уравнения: С 2 К = 1 . При концентрации кислоты или основания С и степени диссоциации α, концентрация ионов водорода и гидроксила составит: СН+ = Сα и СОН- = Сα. С учётом этого: С ОН СН Кк = 1 и Кос = 1 . (17) Для слабых кислот и оснований, когда α 1, уравнение (17) принимает вид: Кк = СН+α и Кос = СОН-α. (18) ДИССОЦИАЦИЯ ВОДЫ. рН РАСТВОРОВ. Диссоциация воды протекает по схеме: Н2О Н+ + ОНКонстанту диссоциации можно выразить как KH 2 O = aH aOH aH 2 O Т.к. степень диссоциации воды очень мала, то aH O можно считать постоянной и ввести ее в значение константы диссоциации: KH 2 O aH 2 O Kw aH aOH При Т = const Kw = const и не зависит от концентрации ионов Н+ и ОН-; Kw называется ионным произведением воды. При 25оС Kw = 10-14. В нейтральном растворе aH aOH Kw = 10-7 моль/л. lg CH + lgCOH = -14. 2 рН = lg aH водородный показатель, введенный Зеренсеном (или Соренсен) (1909). рН + рОН = 14. рН = 7 отвечает нейтральному раствору только при 25оС. СН+ = СОН- = 10-7 г-ион/л. Kw очень сильно зависит от Т (увеличивается в 100 раз в интервале 20-100оС), т.е. с ростом Т концентрация образующихся ионов Н+ и ОН- будет увеличиваться, и при t 25оС рН = 7 будет соответствовать кислому раствору, а при t 25оС щелочному. Известно, что при рН раствора равным 7 этот раствор нейтрален, при меньших числах увеличивается кислотность, при больших - щёлочность раствора. Но эти значения справедливы для комнатной температуры в +25 градусов по Цельсию. Если температура раствора возрастает, то увеличивается способность раствора к диссоциации (увеличивается константа диссоциации), что приводит к тому, что нейтральная реакция будет при значениях рН меньших 7, это обусловлено большими концентрациями положительных Н и отрицательных ОН ионов. Кислые растворы такие, в которых Н+ ОН-; щелочные ОН- Н+. Т.к. мерой кислотности служит Н+, то в ряду кислот более сильной будет та, у которой при одинаковой аналитической концентрации Н+ выше (т.е. больше степень диссоциации). Следует помнить, что величины концентраций ионов Н+ в выражении для рН можно использовать взамен активностей только в случае достаточно разбавленных растворов. БУФЕРНЫЕ РАСТВОРЫ. Буферными растворами называются растворы с устойчивой активностью водородных ионов, т.е. с определенным значением рН, которое практически не зависит от разбавления раствора и слабо меняется при прибавлении к раствору сильной кислоты или сильного основания эта способность и называется буферностью. Обычно буферные растворы представляют собой растворы, содержащие слабую кислоту (или основание) вместе с ее солью. Пример смесь уксусной кислоты и ацетата Na. В растворе имеется сопряженная пара СН3СООН и СН3СОО-, действие которой состоит в том, что при добавлении сильной кислоты ее протоны связываются основанием СН3СОО-, а при введении сильного основания уксусная кислота отдает ему свои протоны, и в обоих случаях Н+ изменяется незначительно. Найдем количественную связь между значением рН и активностями (в случае разбавленных растворов концентрациями) растворенных веществ: Кд,к = aCH COO aH O 3 3 aCH 3COOH ; aH O = Кд,к 3 a CH 3 COOH aCH COO 3 Если к раствору слабой кислоты добавлена соль этой кислоты и сильного основания, то диссоциация кислоты подавляется, и aCH COOH равна общей активности кислоты ак (в растворе смеси уксусной кислоты и ацетата натрия), а aCH COO можно 3 3 считать равной активности ацетата натрия ас : aH O = Кд,к 3 ak ac Подбирая различные отношения концентраций кислоты и соли, можно получить растворы с различными значениями рН. При данном составе раствора значение рН мало зависит от Т. Другой пример буферного раствора слабое основание NH4OH и его соль с сильной кислотой NH4Cl. Для этой сопряженной пары получим: Кд,о = aOH = Кд,о ao ac a NH aOH 4 a NH 4 OH ; = aOH aH O = 3 Kw aOH ac ao = Kw ac Kд ,о ao Анализ уравнений показывает, почему разбавление буферных растворов практически не меняет рН растворов : рН зависит не от абсолютных значений концентраций кислоты и ее соли, а от их соотношения. Количественной характеристикой буферности является буферная емкость число эквивалентов щелочи или кислоты, необходимое для изменения рН на 1: = m pH Величина зависит от Кд,к ( Кд,о) и максимальна в области рН, близкой к рК. Буферные емкости одного и того же раствора относительно кислоты и щелочи различны. Буферные смеси можно приготовить не только из кислоты (или основания) и соли, но и из двух солей на разных ступенях диссоциации (Na2HPO4 и NaH2PO4). Буферные смеси находят применение при измерениях рН растворов, для проведения химических процессов в условиях постоянства рН. 11. Диссоциация слабых электролитов. Константа диссоциации. Степень диссоциации. ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ. При диссоциации слабых электролитов устанавливается равновесие между недиссоциированными молекулами и ионами. Рассмотрим простейший пример, когда молекула распадается только на два иона: СН3СООН + Н2О СН3СОО- + Н3О+ На основании закона действующих масс напишем выражение для константы равновесия Ка: aCH COO aH O 3 3 Ка = aCH 3COOH aH 2 O Активность растворителя (воды) в разбавленных растворах можно считать постоянной: Ка a H O = Кд = aCH COO aH O 2 3 3 aCH 3COOH Величина Кд называется термодинамической константой диссоциации (или просто константой диссоциации). Ка зависит от Т, но не зависит от концентрации растворенного вещества. Заменив активности произведениями аналитических концентраций на соответствующие коэффициенты активности, получим, например, пользуясь моляльностью: Кд = mCH COO mH O 3 3 mCH 3COOH CH COO H O 3 3 CH COOH = Кm CH COO H O 3 3 CH COOH 3 3 Можно выразить Кд через молярность с и соответствующие коэффициенты активности: Кд = cCH COO cH O 3 cCH 3COOH 3 f CH COO f H O 3 f CH 3 COOH 3 = Кс f CH COO f H O 3 3 f CH 3 COOH Кс классическая константа диссоциации. Для точных расчетов ионных равновесий необходимо пользоваться термодинамической константой диссоциации Кд. Коэффициенты активности можно рассчитать по уравнениям Дебая-Гюккеля. 12. Ионное равновесие в растворах электролитов. Гидролиз. ГИДРОЛИЗ СОЛЕЙ. Гидролизом называется обменная реакция вещества с водой. В результате гидролиза солей сильных кислот и слабых оснований (NH4Cl) или слабых кислот и сильных оснований (СН3СООNa) образуются слабодиссоциированные кислоты или основания, что приводит к нарушению равенства концентраций ионов Н + и ОН- в растворе и раствор приобретает кислую или щелочную реакцию. СН3СОО- + Н2О СН3СООН + ОНКа = aCH 3COOH aOH aCH COO aH 2 O 3 В разбавленных растворах aH O можно считать постоянной: 2 Ка a H O = Кг = aCH 3COOH aOH aCH COO 2 3 Кг константа гидролиза. Константа диссоциации уксусной кислоты: Кд,к = aCH COO aH O 3 3 aCH 3COOH = aCH COO aH O / Кд,к ; aCH 3COOH 3 3 После подстановки (учитывая, что aOH aH O = Кw): 3 Кг = Кw / Кд,к Аналогично для соли сильной кислоты и слабого основания (NH4Cl) можно получить: Кг = Кw / Кд,о Для соли слабой кислоты и слабого основания: CH3COO- + NH4+ + H2O CH3COOH + NH4OH Кг = aCH 3 COOH a NH 4 OH aCH COO a NH 3 aCH 3COOH = aCH COO aH O / Кд,к 3 3 4 a NH 4 OH = aNH aOH / Кд,о ; 4 Кг = aOH aH O / Кд,к Кд,о = Кw / Кд,к Кд,о 3 Т.о., константы гидролиза солей можно рассчитать, зная константы диссоциации соответствующих слабых кислот и оснований. Наряду с константой гидролиза реакции гидролиза характеризуются также степенью гидролиза. Степень гидролиза доля молекул соли, подвергшихся гидролизу, от общего числа молекул соли. Для гидролиза CH3COONa: = OH CH COONa 3 c 2 c c Кг = = 1 c 1 где с исходная концентрация CH3COONa. В случае гидролиза соли слабого основания и сильной кислоты связь между Кг и точно такая же. Зная Кг, можно рассчитать , решая уравнение: с2 + Кг Кг = 0 При малых значениях ( 1 и 1 1) Кг = с2 и = Kг / с . Если соль образована слабой кислотой и слабым основанием, 2 c c Кг = 2 = 2 c 1 1 2 При 1 Кг = 2 и = Kг . Анализ уравнений показывает, что степень гидролиза солей слабых кислот и сильных оснований и солей сильных кислот и слабых оснований уменьшается с ростом концентрации соли и увеличивается при разбавлении растворов; степень гидролиза солей слабого основания и слабой кислоты от концентрации раствора практически не зависит. Зависимость Кг от температуры: Кw возрастает с ростом Т очень значительно, Кд от Т почти не зависит, следовательно, Кг с повышением Т увеличивается, соответственно возрастает и . Гидролиз солей можно представить, как поляризационное взаимодействие ионов и их гидратной оболочки. Гидролиз протекает тем полнее, сильнее поляризующее действие ионов. Возможны 4 случая протекания гидролиза: 1. Соли, образованные сильным основанием и сильной кислотой. В этом случае, гидролиз практически не происходит, т.к. катионы и анионы, образующиеся в растворе при диссоциации соли, слабо поляризуют гидратную оболочку. pH среды не изменяется (рН ≈ 7): NaCl ↔ Na+ + Cl— Na+ + HOH ↔ реакция практически не протекает Cl— + HOH ↔ реакция практически не протекает 2. Соли, образованные слабым основанием и сильной кислотой (гидролиз по катиону) Такое соединение, при ионизации, образует катионы, способные к поляризации гидратной оболочки и анионы, которые их поляризуют слабо. Тогда гидролиз проходит по катиону, при этом среда носит кислый характер, т.е. рН ˂ 7: NH4Cl ↔ NH4+ + Cl— NH4+ + HOH ↔ NH4OH + H+ Cl—+ HOH ↔ реакция практически не идет NH4Cl+ HOH ↔ NH4OH + HCl Для солей, образованных слабым основанием и сильной кислотой, константа гидролиза и константа диссоциации основания связаны соотношением: Kг = KH2O/Kосн Понятно, что чем меньше сила основания, тем в большей степени протекает гидролиз. Если соль образованна слабым основанием многовалентного металла и сильной кислотой, то ее гидролиз будет протекать ступенчато: FeCl2 ↔ Fe2+ + 2Cl— Fe2++HOH↔(FeOH)+ +H+ I ступень FeCl2 + HOH ↔ (FeOH)Cl + HCl (FeOH)+ +HOH↔Fe(OH)2 +H+ II ступень (FeOH)Cl + HOH↔ Fe(OH)2 + HCl Константа гидролиза по первой ступени связана с константой диссоциации основания по второй ступени, а константа гидролиза по второй ступени — с константой диссоциации основания по первой ступени: Kг1 = KH2O/Kосн2, Kг2 = KH2O/Kосн1 Поскольку первая константа диссоциации кислоты всегда больше второй, то первая константа гидролиза всегда больше, чем константа вторая гидролиза, так как первая константа диссоциации основания всегда больше второй Kг1 > Kг2 Отсюда следует, что по первой ступени, гидролиз всегда будет протекать в большей степени, чем по второй. Этому также способствуют ионы, которые образуются при гидролизе по первой ступени, они приводят к подавлению гидролиза по второй ступени, смещая равновесие влево. Сравнивая величины Kг и Kосн можно качественно определить pH среды. Так, если Kг намного больше Kосн, то среда сильнокислая, при Kг намного меньшей Kосн — среда слабокислая, а если Kги Kосн сопоставимы, то — среднекислая. 3. Соль, образованная сильным основанием и слабой кислотой (гидролиз по аниону) Такое соединение в растворе образует слабополяризующие катионы и среднеполяризующие анионы. Гидролиз протекает по аниону, и в его результате создается щелочная среда, pH> 7: NaCN ↔ Na+ + CN— CN— + HOH ↔ HCN + OH— Na+ + HOH ↔ реакция практически не идет NaCN + HOH ↔ HCN + NaOH Константа гидролиза и константа диссоциации кислоты связаны зависимостью: Kг = KH2O/Kк-ты Т.е. гидролиз соли протекает тем полнее, чем слабее образующая эту соль, кислота. Возможен гидролиз соли, образованной слабой многоосновной кислотой и сильным основанием. В этом случае гидролиз протекает по ступеням: Na2SO3 ↔ 2Na+ + SO32SO32-+HOH↔HSO3—+OH— I ступень Na2SO3+HOH↔NaHSO3+NaOH HSO3—+HOH↔H2SO3 +OH— II ступень NaHSO3 + HOH ↔ H2SO3 + NaOH В этом случае, константа гидролиза по первой и второй ступеням определяется соотношениями: Kг1 = KH2O/Kк-ты2, Kг2 = KH2O/Kк-ты1 Следует помнить, что гидролиз по второй ступени протекает в ничтожно малой степени. Сравнивая величины Kг и Kк-ты, можно качественно определить pH среды. Так, если Kг намного больше Kк-ты, то среда сильнощелочная, при Kг намного меньшей Kк-ты — среда слабощелочная, а если Kги Kосн сопоставимы, то — среднещелочная. 4. Соли, образованные слабым основанием и слабой кислотой. Такие соли, при ионизации образуют среднеполяризующие катионы и анионы, поэтому гидролиз возможен как по катиону, так и по аниону. При этом относительная сила образовавшихся кислоты и основания, будут влиять на характер среды (слабокислая или слабощелочная, pH ≈ 7). Такого типа гидролиз протекает особо полно, обычно с образованием малорастворимого вещества: Al2S3 + 6HOH ↔ 2Al(OH)3↓+ 3H2S↑ Константу гидролиза можно рассчитать, зная константы диссоциации кислоты и основания с помощью следующего соотношения: Kг = KH2O/(Kк-ты·Kосн) Совместный гидролиз протекает при взаимодействии растворов двух солей, одна из которых образована слабым основанием и сильной кислотой, а вторая напротив сильным основанием и слабой кислотой. Т.е. одна соль гидролизуется по катиону, а другая – по аниону. В таких случаях гидролиз взаимно усиливается. Например, рассмотрим совместный гидролиз растворов солей хлорида алюминия и сульфида натрия: При гидролизе хлорида алюминия соль гидролизуется по катиону: AlCl3 ↔ Al3+ + 3Cl— Al3+ + 3HOH ↔ Al(OH)3 + 3H+ При гидролизе сульфида натрия соль гидролизуется по аниону: Na2S ↔ 2Na+ + S2S2- + 2HOH ↔ H2S + 2OH— Суммарная реакция гидролиза: 2AlCl3 + 3Na2S + 6H2O = 2Al(OH)3↓ + 3H2S↑ + 6NaCl Влияние различных факторов на протекание гидролиза Природа соли. Это видно из выражения для константы гидролиза. Концентрация соли и продуктов реакции. В соответствии с принципом Ле-Шателье, равновесие должно смещаться вправо, при этом увеличивается концентрация ионов водорода (или гидроксид-ионов), что приводит к уменьшению степени гидролиза. Температура. Известно, что гидролиз притекает с поглощением теплоты (эндотермическая реакция), поэтому согласно принципу Ле Шателье, при увеличении температуры равновесие сдвигается вправо, что ведет к росту степени гидролиза. 13. Ионное равновесие в растворах электролитов. Произведение растворимости. ПРОИЗВЕДЕНИЕ РАСТВОРИМОСТИ. При растворении твердых веществ в воде может быть достигнуто состояние насыщения раствора, при котором для данной Т растворение вещества прекратится, при этом твердая фаза будет находиться в равновесии с растворенным веществом. Если вещество (соль MnAm) является электролитом, то в растворе установится равновесие MnAm (тв.) n M+ + m Aa Mn a Am Константа диссоциации соли Кд = ; a M n Am при данной Т является a M n Am величиной постоянной. Кд a M n Am = ПР = a M a A Т.о., в насыщенном растворе соли произведение активностей ее ионов есть величина постоянная, она называется произведением растворимости. Если соль малорастворима и ее раствор разбавлен, то в выражении для ПР активности можно заменить концентрациями. Из уравнения для ПР следует, что при добавлении к раствору малорастворимой соли хорошо растворимого соединения, имеющего общий ион с малорастворимой солью, растворимость соли будет уменьшаться. Кроме того, добавление посторонней соли к раствору увеличивает ионную силу этого раствора и уменьшает коэффициент активности той соли, которая находилась в растворе до введения посторонней добавки. Величины ПР имеют большое практическое значение в химической технологии и в аналитической химии, т.к. они определяют условия, при которых должно происходить растворение солей или выделение их из растворов. ПР наиболее точно можно измерить методом ЭДС; часто также пользуются определением растворимости по электропроводности насыщенных растворов, однако этот метод применим только к растворам чистой соли. n m Произведение активностей (ПА), или произведение термодинамических активностей ионов в насыщ. р-ре электролита (соль, гидроксид и т. п.) в данном р-рителе. AkBl(тв) kAz+ (р-р) + lВz- (р-р). По закону действующих масс, константа равновесия где аАz+, аBz-, аАkBl - активности катионов, анионов и растворенного в-ва. Т. к. в насыщ. р-р с аАkBl =1. ПА для ионов ПА зависит от температуры и природы растворителя, а при фиксир. т-ре в данном р-рителе ПА для каждой соли - пост. величина, характеризующая ее растворимость. Если в р-ре произведение > ПА, твердое соед. АkВl выпадает в осадок, если же - <ПА, в-во АkВl переходит в р-р. В аналит. химии вместо строгого термодинамич. понятия ПА используется произведение растворимости (ПР), равное произведению молярных концентраций ионов [Az+]k и [Вz-]l в р-ре соли АkВl. ПР связано с ПА через коэф. активности и ПА = ПР. В реальном р-ре ПР, в отличие от ПА, не является пост. величиной: для данного р-рителя при пост. т-ре (т.е. при ПА = const) присутствие в р-ре других растворенных в-в, помимо АkВl , или изменение концентраций ионов Az+ и Вz- приводит к изменению коэф. активности и, следовательно, к изменению ПР. На практике обычно считают р-ры идеальными, т. е. принимают ПА = ПР = const. ПР связано с р-римостью S (обычно измеряемой в моль/л) соотношением: Для осаждения соли АkВl из ненасыщ. р-ра обычно добавляют в р-р соль, имеющую с ней общий катион или анион, AD или СВ. При этом концентрация ионов Аz+ (или Вz-) в р-ре возрастает, произведение концентраций [Az+]k[Bz-]l превышает ПР и начинается выпадение соли в осадок. Наоборот, полный переход АkВl в р-р достигается связыванием одного из ионов z+ (А или Bz-) в недиссоциируемый комплекс в р-ре, что приводит к снижению концентрации этого иона и выполнению условия [Az+]k[Bz-]l< ПР. Третий способ регулирования р-римости - изменение ионной силы р-ра (т. наз. солевой эффект), когда к р-ру добавляется электролит CD, не имеющий общих ионов с АkВl. Это приводит к росту ионной силы, уменьшению a и, следовательно, увеличению р-римости. Образование осадков [Ag+] [Cl-] < ПРAgCl - ненасыщенный раствор [Ag+] [Cl-] = ПРAgCl - насыщенный раствор [Ag+] [Cl-] > ПРAgCl - перенасыщенный раствор Осадок образуется в том случае, когда произведение концентраций ионов малорастворимого электролита превысит величину его произведения растворимости при данной температуре. Когда ионное произведение станет равным величине ПР, выпадение осадка прекращается. Влияние концентрации растворов Труднорастворимый электролит с достаточно большой величиной ПР нельзя осадить из разбавленных растворов. Например, осадок PbCl2 не будет выпадать при смешении равных объемов 0,1 M растворов Pb(NO3)2 и NaCl. При смешивании равных объемов концентрации каждого из веществ станут 0,1 / 2 = 0,05 M или 5 10-2 моль/л. Ионное произведение [Pb2+] [Cl1-]2 = 5 10-2 (5 10-2)2 = 12,5 10-5. Полученная величина меньше ПРPbCl2, следовательно выпадения осадка не произойдет. Влияние количества осадителя Для возможно более полного осаждения употребляют избыток осадителя. Например, осаждаем соль BaCO3: BaCl2 + Na2CO3 ® BaCO3¯ + 2NaCl. После прибавления эквивалентного количества Na2CO3 в растворе остаются ионы Ba2+, концентрация которых обусловлена величиной ПР. Повышение концентрации ионов CO32-, вызванное прибавлением избытка осадителя (Na2CO3), повлечет за собой соответственное уменьшение концентрации ионов Ba2+ в растворе, т.е. увеличит полноту осаждения этого иона. Влияние одноименного иона Растворимость труднорастворимых электролитов понижается в присутствии других сильных электролитов, имеющих одноименные ионы. Если к ненасыщенному раствору BaSO4 понемногу прибавлять раствор Na2SO4, то ионное произведение, которое было сначала меньше ПРBaSO4 (1,1 10-10), постепенно достигнет ПР и превысит его. Начнется выпадение осадка. Влияние температуры ПР является постоянной величиной при постоянной температуре. С увеличением температуры ПР возрастает, поэтому осаждение лучше проводить из охлажденных растворов. Растворение осадков Предположим, что надо растворить осадок BaСO3. Раствор, соприкасающийся с этим осадком, насыщен относительно BaСO3. Это означает, что [Ba2+] [CO32-] = ПРBaCO3. Если добавить в раствор кислоту, то ионы H+ свяжут имеющиеся в растворе ионы CO32- в молекулы непрочной угольной кислоты: 2H+ + CO32- ® H2CO3 ® H2O + CO2 Вследствие этого резко снизится концентрация иона CO32- , ионное произведение станет меньше величины ПРBaCO3. Раствор окажется ненасыщенным относительно BaСO3 и часть осадка BaСO3 перейдет в раствор. При добавлении достаточного количества кислоты можно весь осадок перевести в раствор. Следовательно, растворение осадка начинается тогда, когда по какой-либо причине ионное произведение малорастворимого электролита становится меньше величины ПР. Для того, чтобы растворить осадок, в раствор вводят такой электролит, ионы которого могут образовывать малодиссоциированное соединение с одним из ионов труднорастворимого электролита. Этим объясняется растворение труднорастворимых гидроксидов в кислотах В реальных условиях анализа сравнительно редко приходится иметь дело с насыщенными растворами малорастворимых соединений, не содержащими каких-либо посторонних ионов, которые способны взаимодействовать с ионами осадка. Эти конкурирующие реакции приводят к увеличению растворимости. Правило постоянства произведения концентраций следует из применения закона действия масс к насыщенному раствору малорастворимого электролита. Однако это правило имеет приближенный характер, потому что равновесие между осадком и раствором характеризуется более сложной зависимостью. При введении в насыщенный раствор труднорастворимой соли постороннего электролита состояние равновесия нарушается, часть твердой фазы будет переходить в раствор и растворимость осадка увеличится. В присутствии посторонних электролитов коэффициенты активности ионов, которые зависят от ионной силы раствора, всегда меньше единицы. Отсюда можно заключить, что произведение растворимости, а также и растворимость малорастворимых соединений увеличивается в растворах с повышением концентрации сильных электролитов. Процесс растворения твердой фазы проходит до тех пор, пока активность ионов в растворе, т. е. их способность к взаимным столкновениям, не станет такой же, как и до введения в раствор постороннего электролита. После этого снова установится динамическое равновесие между осадком и ионами раствора. Поэтому постоянной величиной является не произведение концентрации ионов, а произведение их активности.