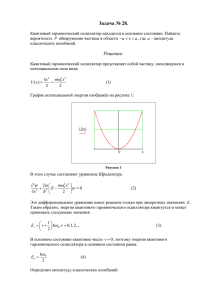
термодинамика и статистическая физика - Сайт 1
реклама

БЕЛОРУССКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ Факультет радиофизики и оптоэлектроники Бортник Михаил Васильевич Ушаков Дмитрий Владимирович ТЕРМОДИНАМИКА И СТАТИСТИЧЕСКАЯ ФИЗИКА (Краткий конспект лекций) Минск 2002 г. Содержание 1 РАВНОВЕСНАЯ ФЕНОМЕНОЛОГИЧЕСКАЯ ТЕРМОДИНАМИКА 1.1 Начала термодинамики . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.2 Калорическое и термические уравнения состояния. Число степеней свободы термодинамической системы . . . . . . . . . . . . . . . . . . . . . . 1.3 Методы равновесной термодинамики . . . . . . . . . . . . . . . . . . . . 1.4 Термодинамические потенциалы для закрытой системы . . . . . . . . . . 1.5 Термодинамические потенциалы для открытой системы . . . . . . . . . . 1.6 Общие и частные условия равновесия и устойчивости термодинамических систем . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.7 Дополнительные вопросы . . . . . . . . . . . . . . . . . . . . . . . . . . . 8 10 2 Равновесные функции распределения (Гиббса) термодинамических систем 2.1 Энтропия и вероятность . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2.2 Принцип равной вероятности. Микроканоническое распределение . . . . 2.3 Каноническое распределение . . . . . . . . . . . . . . . . . . . . . . . . . 2.4 Обобщенное распределение . . . . . . . . . . . . . . . . . . . . . . . . . 2.5 Связь квантовых и классических распределений . . . . . . . . . . . . . . 2.6 Дополнительные вопросы . . . . . . . . . . . . . . . . . . . . . . . . . . . 11 11 13 14 16 18 19 3 Статистическая термодинамика равновесных систем 3.1 Статистическое представление внутренней энергии . . . . . . . . . . . . 3.2 Статистическое представление числа частиц . . . . . . . . . . . . . . . . 3.3 Свободная энергия (F) и омега-потенциал (Ω) . . . . . . . . . . . . . . . 3.4 Методы статистической термодинамики . . . . . . . . . . . . . . . . . . . 3.5 Статистическое обоснование начал феноменологической термодинамики 3.6 Дополнительные вопросы . . . . . . . . . . . . . . . . . . . . . . . . . . . 20 20 20 21 22 23 23 4 Идеальные ТД системы в равновесном состоянии. Статистика Больцмана 4.1 Классическое распределение Максвелла-Больцмана . . . . . . . . . . . 4.1.1 Приложение . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4.2 Термодинамические свойства классического газа "бесструктурных"частиц 4.3 Одночастичное распределение Больцмана для систем с дискретным спектром состояний . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4.4 Термодинамические свойства квантового больцмановского газа "бесструктурных"частиц . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4.5 Статистические суммы системы квантовых осцилляторов и квантовых ротаторов . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4.6 Распределение Больцмана для чисел заполнения . . . . . . . . . . . . . . 4.7 Больцмановский газ с двумя энергетическими уровнями . . . . . . . . . . 4.8 Термодинамические свойства молекулярного газа. Квантовоклассическое распределение Больцмана . . . . . . . . . . . . . . . . . . 4.9 Отрицательные абсолютные температуры . . . . . . . . . . . . . . . . . . 4.10 Дополнительные вопросы . . . . . . . . . . . . . . . . . . . . . . . . . . . 24 24 26 28 2 3 3 4 4 6 7 31 33 34 36 37 38 42 43 5 Идеальные системы в равновесном состоянии. Распределение Ферми. Распределение Бозе 5.1 Обобщенное распределение (Гиббса) для системы однотипных частиц . . 5.2 Распределение Ферми. Распределение Бозе . . . . . . . . . . . . . . . . 5.3 Критерий применения статистики Больцмана . . . . . . . . . . . . . . . . 5.4 Термодинамические свойства газа ферми-частиц . . . . . . . . . . . . . . 5.5 Термодинамические свойства газа бозе-частиц . . . . . . . . . . . . . . . 5.6 Статистика электронов в металлах и полупроводниках . . . . . . . . . . 5.7 Равновесное тепловое излучение . . . . . . . . . . . . . . . . . . . . . . . 5.8 Метод наиболее вероятного распределения . . . . . . . . . . . . . . . . . 5.9 Дополнительные вопросы . . . . . . . . . . . . . . . . . . . . . . . . . . . 44 44 46 46 47 50 52 55 57 59 6 Равновесные ТД системы взаимодействующих частиц 6.1 Конфигурационный интеграл. Частичные функции распределения . . . . 6.2 Термическое уравнение состояния газовой среды с парным взаимодействием . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6.3 Термодинамические свойства кристаллических тел . . . . . . . . . . . . . 6.4 Термодинамические свойства классической невырожденной плазмы . . . 6.5 Дополнительные вопросы . . . . . . . . . . . . . . . . . . . . . . . . . . . 60 61 7 Равновесные флуктуации 7.1 Флуктуации энергии . . . . . . . . . . . . . . . . . . 7.2 Флуктуации числа частиц . . . . . . . . . . . . . . . 7.3 Эквивалентность функций распределений Гиббса . 7.4 Распределение вероятности для флуктуаций . . . . 7.5 Флуктуация основных термодинамических величин 7.6 Дополнительные вопросы . . . . . . . . . . . . . . . 69 69 70 70 71 72 73 3 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63 64 66 68 1 РАВНОВЕСНАЯ ФЕНОМЕНОЛОГИЧЕСКАЯ ТЕРМОДИНАМИКА Теория феноменологической термодинамики в полном объеме представлена в литературе: И.П.Базаров, Термодинамика, М., Высшая школа, 1983; М.А.Леонтович, Введение в термодинамику. Статистическая физика, М., Наука, 1983. В данной части мы ограничимся рассмотрением только тех вопросов, которые необходимы при обсуждении методов статистической термодинамики, позволяющей в наиболее общей форме проводить изучение свойств макроскопических объектов (см. Часть 3). 1.1 Начала термодинамики Термодинамика - дедуктивная дисциплина. Она позволяет получить множество различных соотношений между величинами, характеризующими различные объекты, опираясь на общие эмпирические законы - начала термодинамики. Первое начало. Определяет внутреннюю энергию (E) системы как однозначную функцию ее состояния. Изменение E представляется в виде: dE = dQ − dW + dZ (1) dE = dQ − dW (2) - для открытой системы, - для закрытой системы, dE = 0, E = const (3) - для изолированной системы, где dQ- элементарное количество теплоты, dW - элементарная работа, dZ- элементарное количество энергии, связанной с переносом вещества. Второе начало. Определяет энтропию (S) системы как однозначную функцию ее состояния. Ее изменение представляется в виде: dQ , (4) T где T - абсолютная термодинамическая температура. Третье начало. Определяется свойство энтропии S в области низких температур ∂S lim = 0, (5) T →0 K ∂X T dS = где X - любой термодинамический параметр. Объединение 1-го и 2-го начал термодинамики приводит к основному термодинамическому равенству (6) dE = T dS − Ada + µdN, где использованы представления dW = Ada, при этом A - сопряженная внешнему параметру a обобщенная сила; dZ = µdN, при этом µ - химический потенциал переносимых частиц dN. В общем виде основное термодинамическое равенство определяется выражением dE = T dS − ∑ µ j dN j . j 4 (7) 1.2 Калорическое и термические уравнения состояния. Число степеней свободы термодинамической системы Уравнения состояния представляют термодинамическую систему. Для их определения используют нулевое (первое) начало термодинамики (8) bi = bi (a1 , a2 , ..., an , T ) , где bi - любая внутренняя величина системы, a j - внешняя величина системы. В связи с этим, под калорическим уравнением состояния для закрытой системы принимают функциональную связь (9) E = E (a1 , a2 , ..., T ) . Термические уравнения состояния определяются выражением (10) A j = A j (a1 , a2 , ..., T ) Таким образом, в простейшем случае закрытой системы уравнения состояния представляются в виде E = E (a, T ) и A = A (a, T ). Предполагается, что в рамках непосредственно теории термодинамики уравнения состояния определяются экспериментально. В связи с этим теорию в целом характеризуют как феноменологическую. Рассмотрим представление числа степеней свободы системы. Допустим, имеем простейший случай для системы dE = T dS − Ada E = E (a, T ) , A = A (a, T ) . (11) Тогда из пяти величин, представленных в основном термодинамическом равенстве, две величины следует рассматривать как параметры системы, т.к. они не могут быть установлены из представленной системы уравнений. Эти параметры следует рассматривать как независимые величины и их число определяет число степеней свободы термодинамической системы. Наиболее целесообразно в качестве независимых в данном случае выбрать a и T . Однако, как будет показано далее, теория термодинамики настолько формализована, что в качестве независимых переменных можно выбрать любую пару величин, присутствующих в основном термодинамическом равенстве. Понятие числа степеней свободы легко обобщается и на более общие случаи. 1.3 Методы равновесной термодинамики Рассмотрим ряд выражений, которые можно получить используя 1-ое и 2-ое начало термодинамики. Допустим, имеем dQ = dE + Ada. (12) Выберем в качестве пары независимых переменных a и T . Тогда из первого начала получаем: dE + Ada 1 ∂E 1 ∂E dS = = dT + +A (13) T T ∂T a T ∂a T 5 Тогда ∂S ∂T 1 = T ∂E ∂T , a ∂S ∂a 1 = T ∂E ∂a +A . (14) T Используя условие полного дифференциала для S, получаем ∂A ∂E = + A. T ∂T a ∂a T (15) Запишем выражения для калорических и термических коэффициентов. Теплоемкости Ca и CA представляются выражениями dQ dE Ca = = , dT a dT a (16) dQ ∂E ∂E ∂A CA = = + +A dT A ∂T a ∂A T ∂T A Если использовать второе начало термодинамики, то получаем ∂A ∂a CA −Ca = T . ∂T a ∂T A (17) Установим связь между калорическими коэффициентами (Cv , C p ) и термическими коэффициентами 1 ∂v α= (18) v0 ∂T p – объемное расширение, 1 β=− v0 ∂v ∂T (19) T – термическое сжатие. Используя тождество ∂v ∂T ∂p = −1 ∂T P ∂p v ∂v T (20) и выражения для α (18) и β (19) получаем ∂p ∂v ∂p α =− = . ∂T v ∂T p ∂v T β (21) Тогда, используя выше представленное выражение для CA −Ca , имеем C p −CV = V0 T α2 . β (22) Изменение энергии системы представляется выражением Z2 1 где считаем Ca = ∂E ∂T dE = E2 − E1 = Z2 Z2 Ca dT + 1 1 . a 6 ∂E ∂a da, T (23) Изменение энтропии системы представляется выражением Z2 1 dS = S2 − S1 = Z2 dT Ca + T Z 1 ∂A ∂T da. (24) a Рассмотренные соотношения могут быть применены к конкретной системе, если она представлена уравнениями состояний. 1.4 Термодинамические потенциалы для закрытой системы Рассмотрим основное термодинамическое равенство в виде dE = T dS − Ada. (25) 1. Выберем в качестве пары независимых переменных S и a. Считаем, что известно уравнение E = E (S, a). Тогда для остальных величин имеем ∂E ∂E T= , A=− . (26) ∂S a ∂a S Из условия полного дифференциала для E записываем ∂T ∂A =− . ∂a S ∂S a (27) Вторые производные от E (S, a) позволяют определить калорические и термические коэффициенты 2 ∂ E ∂T T = = , 2 ∂S ∂S C a a a 2 , (28) ∂ E ∂P KS =− = , ∂V 2 S ∂V S V ∂P где KS = −V - адиабатный модуль упругости. Функциональную связь E = E (S, a) ∂V S - называют термодинамическим потенциалом внутренняя энергия. 2. Термодинамический потенциал свободная энергия F определяется выражением F = E − T S, дифференциал для которой представляется в виде dF = −SdT − Ada, т.е. имеем F = F (a, T ). Это выражение, также позволяет получить целый ряд термодинамических соотношений. Действительно сразу получаем ∂F ∂F S=− , A=− . (29) ∂T a ∂a T Условие полного дифференциала для F приводит к уравнению ∂S ∂A = ∂a T ∂T a (30) Вторые производные от функции F определяют теплоемкость и коэффициент сжатия 2 ∂ F 1 ∂V 1 Ca = −T , β=− = 2 . (31) 2 ∂T a V0 ∂P T ∂ F V0 ∂V 2 T 7 3.Термодиномический потенциал Гиббса представляется функцией вида Φ = (E − T S) + Aa. Дифференциал для Φ имеет вид dΦ = −SdT + adA, т.е. имеем Φ = Φ (T, A). Из этого уравнения получаем ∂Φ ∂Φ S=− , a= . (32) ∂T A ∂A T Вторые производные от Φ = Φ (T, A) дают теплоемкость 2 ∂ Φ CA = −T , ∂T 2 A (33) коэффициент сжимаемости 1 β=− V0 ∂V ∂P T 1 =− V0 ∂2 Φ ∂P2 . (34) T 4. Термодинамический потенциал энтальпия H представляется выражением H = E + Aa. Тогда имеем dH = T dS + adA, т.е. H = H (S, A). В этом случае имеем ∂H ∂H T= , a= , 2 ∂S A 2∂A S ∂ H T ∂ H ∂V V (35) = , = = − , 2 2 ∂S C ∂P ∂P K A S A S S ∂T ∂a = . ∂A S ∂S A Очевидно, что полученные выражения для различных величин имеют общий характер. Конкретная информация получается, если известны функциональные связи E(S, a), F(a, T ), Φ(T, A), H(S, A). Это позволяет считать, что термодинамические потенциалы, наряду с уравнениями состояние (см. п. 1.2), могут также выступать в качестве характеристик, определяющих термодинамические системы. Полагается, что в рамках теории термодинамики аналитические выражения для потенциалов устанавливаются экспериментально 1.5 Термодинамические потенциалы для открытой системы Рассмотрим основное термодинамическое равенство в виде dE = T dS − Ada + µdN. (36) Это выражение (по аналогии с закрытыми системами см. п. 1.4) следует рассматривать как дифференциал термодинамического потенциала - внутренняя энергия с естественными переменами ( S, a, N), т.е. E = E(S, a, N). Для потенциала F - свободная энергия, исходя из определения F = E − T S, в данном случае имеем dF = −SdT − Ada + µdN, (37) где F = F(T, a, N). Для потенциала Φ - Гиббса, исходя из определения Φ = F + Aa, в данном случае получаем dΦ = −SdT + adA + µdN, (38) 8 где Φ = Φ(T, A, N). Для потенциала H - энтальпия, исходя из определения H = E + Aa, в данном случае получаем (39) dH = T dS + adA + µdN, где H = H(S, A, N). Эти выражения позволяют получить множество различных соотношений между величинами, определяющими свойства термодинамических систем. Остановимся на рассмотрении химического потенциала µ (введенное ранее в п. 1.1). Как следует, для µ имеем ∂E ∂F ∂Φ ∂H µ= = = = . (40) ∂N α,s ∂N α,T ∂N A,T ∂N A,S Можно уточнить представление химического потенциала. Для этого будем считать, что термодинамические потенциалы являются аддитивными величинами. Тогда каждый из них представляется в виде E = N f1 (a/N, S/N) , Φ = N f3 (A, T ), F = N f2 (a/N, T ) , Из этой системы получаем µ= ∂Φ ∂N H = N f4 (A, S/N). (41) (42) = f3 (A, T ), A,T т.е. Φ = vN, где v следует рассматривать как термодинамический потенциал Гиббса на одну частицу. Введем дополнительно еще один термодинамический потенциал, дифференциал которого позволяет установить число частиц N в системе. Определим функцию Ω = F − Φ - потенциал омега. Тогда имеем Ω = −SdT − Ada − Ndµ, (43) где при дифференцировании Ω учтено представление Φ = µN. Отсюда получаем S=− dΩ , dT A=− dΩ , da N=− dΩ dµ (44) Таким образом, при известном потенциале Ω = Ω(T, a, µ) получаем выражение для числа частиц N в исследуемой системе. 1.6 Общие и частные условия равновесия и устойчивости термодинамических систем При обсуждении этого вопроса используется: • нулевое (второе) начало термодинамики, согласно которому объект стремится перейти в равновесное состояние; • представление любого процесса в общем случае с помощью основного термодинамического неравенства T dS > dE + Ada − µdN. (45) 9 Это позволяет сформулировать общие условия равновесия и устойчивости системы. 1. Изолированная система ( E, a, N) -постоянные). Тогда из неравенства (45) следует dS > 0. В состоянии равновесия должно выполняться δS = 0, δ2 S < 0. 2. Система при постоянных T , a, N. Неравенство принимает вид dF < −SdT − Ada или dF < 0. В состоянии равновесия должно выполняться δF = 0, δ2 F > 0. 3. Система при постоянных T , A, N. Неравенство приводим к виду Φ < −SdT + adA или dΦ < 0. 4. Система при постоянных S, A, N. Аналогично получаем dH < 0. Соответственно должно выполняться δH = 0 и δ2 H > 0. Аналогичные требования выполняются для остальных термодинамических потенциалов ( E и Ω). Частные условия равновесия и устойчивости получают как следствия, вытекающие из общих условий. Рассмотрим методику получения частных условий равновесия на простейшем примере. Рассмотрим однородную изолированную систему и разделим (мысленно) ее на две подсистемы Записываем для энтропия системы S = S1 + S2 , Si энтропии подсистем, и δS = 0 по общему условию равновесия. Тогда получаем dE1 + A1 da1 − µ1 dN1 dE2 + A2 da2 − µ2 dN2 + = 0. T1 T2 (46) Отсюда, учитывая, что dE1 = −dE2 , da1 = −da2 , dN1 = −dN2 , получаем в равновесном состоянии должно выполняться T1 = T2 , A1 = A2 , µ1 = µ2 . Можно показать, что и в более сложных системах требования условия равновесия сводятся к равенству температур, термодинамических сил и химических потенциалов. Детальный анализ приводится, например, в литературе Базаров И.П. "Термодинамика"в разделе "Равновесие в гетерогенной системе". Рассмотрим метод получения частных условий устойчивого состояния на примере закрытой однородной системы. Выберем в качестве независимых переменных S и Q. Тогда общие условия должны представляться с помощью термодинамического потенциала внутренняя энергия E(S, a). В состоянии равновесия должно выполняться δ2 E > 0. Это неравенство сводится к выполнению требования 2 2 ∂ E ∂2 E ∂ E 2 dS + 2 dSda + da2 > 0 (47) 2 2 ∂S a ∂S∂a ∂a S или ∂2 E ∂S2 2 2 ∂ E ∂ E · − > 0. 2 ∂a ∂S∂a a (48) Последнее выражение можно записать в более простой форме 2 2E ∂ E ∂T ∂T ∂ ∂S2 ∂a∂S ∂S a ∂a S a 2 (49) , ∂ E = ∂ (−A) ∂ (−A) ∂E ∂S∂a ∂S ∂a ∂a2 S a S ∂E ∂E где использовано = T, = −A. ∂S a ∂a S Перейдём в дифференциальном определителе от переменных S, a к переменным a, T ∂ (T, −A) ∂ (T, A) ∂ (T, A) ∂ (a, T ) =− =− · . ∂ (S, a) ∂ (S, a) ∂ (a, T ) ∂ (S, a) 10 (50) Раскрывая правую часть выражения, получаем частное условие для конкретной ситуации ∂A ∂T · < 0. (51) ∂a T ∂S a Если принять для A - давление p, а для внешнего параметра a - объём V , то это условие записывается в виде ∂p ∂T ∂p T · · < 0, (52) ∂V T ∂S V ∂V T CV где CV - теплоёмкость системы. 1.7 Дополнительные вопросы 1. Для закрытой системы записать выражение для элементарного количества теплоты dQ в переменных ( V , T ); ( p, T ); ( p, V ). 2. Записать выражение для дифференциалов термодинамических потенциалов F и Φ системы, основное термодинамическое равенство для которой определяется выражением dE = T dS + ∑ Ai dai . i 3. Термодинамический потенциал Φ некоторой системы имеет вид Φ = aT (1 − lnT ) + bT lnp − cT , где a, b, c - постоянные. Установить термическое и калорическое уравнение состояния для системы. 4. Рассматривая энтропию S как термодинамический потенциал закрытой системы, установите для неё естественные переменные. 5. Убедиться, что в предположении постоянства и конечности энтропии S любой системы при T → 0 (третье начало термодинамики) теплоёмкости CV и C p , а также термические коэффициенты расширения и давления стремятся к нулю в этом пределе. 6. Доказать, что основное термодинамическое равенство для диэлектриков в электрическом поле ~E может быть записано в трёх различных формах a) dE = T dS + ~Ed ~D/4π, b) dE ∗ = T dS + ~Ed ~P, c) dE ∗∗ = T dS − ~Pd ~E. (53) Обсудите функциональный вид уравнений состояний в каждом случае. 7. Выбрав наиболее удобный термодинамический потенциал, установить взаи ∂V мосвязь между объемной магнитострикцией и пьезомагнитным эффектом ~ p,T ∂ H ! ~ ∂M . ∂p ~ H,T 8. Низкие температуры с помощью газовых сред получают при H = Const и S = Const. dT Показать, что для коэффициента η = можно получить удобные для практических dp задач выражения ∂V ∂V T −V T ∂T p ∂T p ηH = , ηS = . (54) Cp Cp 11 9. При адиабатном размагничивании вещества возможно охлаждение, т.е. получение низких температур. Показать, что коэффициент, определяющий этот процесс, представляется выражением ! ~ ∂M T ∂T ~ dT H ηS = = . (55) ~ CH dH S ~ 2 Равновесные функции распределения (Гиббса) термодинамических систем Для физических исследований является характерным моделирование. В связи с этим, термодинамические объекты можно рассматривать как динамическую систему с соответствующей функцией Гамильтона H(X), где X – динамические переменные, определяемые степенями свободы частиц системы. Однако, в отличие от обычных (детерминированных) систем механики, для термодинамических объектов характерно их неполное представление состояния. Это связано с тепловым движением частиц. По этой причине состояние термодинамического объекта следует задавать вероятностно – функцией распределения. Основной задачей данной части является установление конкретных выражений для функций распределения. Функция распределения, представляющая динамический процесс в системе, в общем случае определяется в виде P(A1 ,t1 ; A2 ,t2 ; . . . ), где Ai – значение динамических переменных либо функций от них в момент времени ti . В данной части рассматриваются термодинамические системы в равновесном состоянии, когда все величины не зависят от времени. Это относится и к функции распределения. Будем считать, что равновесное состояние термодинамической системы – это такое состояние, когда функция распределения для неё от времени не зависит, т.е. P(A), где A – любая величина, представляющая систему, и значение которой случайно. Доказательство независимости функции распределения от времени систем, для которых функция Гамильтона постоянна, т.е. не зависит от времени, проводится с помощью теоремы Лиувилля (см. Ландау Л.Д., Лифшиц Е.М. Статистическая физика, М., Наука, 1976.) 2.1 Энтропия и вероятность Рассмотрим основное выражение статистической термодинамики S = k lnW ; где S – энтропия системы, W – термодинамическая вероятность, k – постоянная Больцмана. Для W – принимают число состояний, в которых может находиться система в определённых условиях (условия создаются окружающей средой). Величина W может быть представлена просто числом. Однако, для нас важно общее представление для W в зависимости от свойств объекта и влияния окружающей среды. Действительно, в простейшем случае W = W (A), где A – величина, представляющая объект, и по которой считают число состояний. Для термодинамических систем следует положить W = W (A, a1 , a2 , . . . , T ), где ai – внешний параметр для системы, T – 12 температура. Введение таких дополнительных условий в разные "вероятности"типично для статистической термодинамики. Этим отличается статистика физическая от обычной математической вероятности. Выражение для W в ряде случаев точно устанавливается. Рассмотрим несколько простейших примеров. Допустим, частица массы m находится в объёме V и участвует в тепловом движении. Если состояние такой частицы представлять классически, то имеем Z Z W = dxdydzd px d py d pz = V d px d py d pz . Если состояние частицы представлять энергией, тогда, учитывая E = p2 /2m, получаем для E ≤ E0 – заданной W (E ≤ E0 ) = V Z 3 3 4 4πp2 d p = π (2m) 2 V E02 . 3 Для этой же частицы в квантовом представлении имеем En = h̄2 2 n , 2mL2 где в данном случае V = L, n – квантовое число (n = 1, 2, . . .) и квантовомеханическая кратность вырождения gn = 1. Отсюда W будет определяться числом n, т.е. 1 L W (E ≤ E0 ) = (2mE0 ) 2 . h̄ Представленные выражения следует рассматривать как полное число состояний Wn систем для E ≤ E0 . Однако, это число состояний может быть иным, если ввести дополнительные условия. Действительно, если положить E = E0 либо E = E0 ± ∆E, то в этом случае будем получать W < Wn , где W – доступное число состояний в данных условиях. Именно такая вероятность W фигурирует в выражении для энтропии S. Термодинамическую вероятность W следует рассматривать как величину, представляющую состояние системы. Однако, непосредственно она в математическом аппарате статистической термодинамики фактически не используется. Можно показать, что lnW обладает свойствами, характерными для энтропии S системы. Тогда мы придём к формуле, представленной в начале параграфа. При обсуждении этого вопроса будем использовать подход, предложенный в литературе Ф.Рейф, Статистическая физика, М., Наука, 1988. Рассмотрим две макроскопические системы C1 и C2 , которым приписываем энергию E1 и E2 соответственно. Будем считать, что энергия принимает дискретные значения, разделённые интервалами δE. Однако этот интервал достаточно велик, чтобы в нём уместилось большое число состояний. Обозначим через W1 (E1 ) число состояний 1 в интервале энергий E1 + δE1 , а через W2 (E2 ) – число состояний 2 в интервале энергий E2 + δE2 . Считаем, что системы находятся в тепловом контакте, а внешние параметры фиксированы. Полная энергия составной системы : E = E1 + E2 постоянна, и в целом она находится в равновесии. Какова вероятность P(E1 ) того, что энергия системы 1 равна данному значению энергии E1 , т.е. лежит между E и E + δE? Её определим выражением P(E1 ) = WC (E1 ) = Const ·WC (E1 ) WC 13 где Wc – полное число доступных состояний для , когда энергия подсистемы 1 равна E1 . При записи этого выражения использовали принцип равной вероятности. Величину W(1 ) можно выразить через число состояний, доступных системам C1 и C2 WC (E1 ) = W1 (E1 ) ·W2 (E2 ) = W1 (E1 ) ·W2 (E − E1 ). Тогда вероятность того, что система C1 имеет энергию E1 равна P(E1 ) = ConstW1 (E1 )W2 (E − E1 ). Для большинства реальных систем W1 (E1 ) и W2 (E2 ) быстро возрастающие функции энергии. Тогда выражения для P(E1 ) и W1 (E1 ) – быстро возрастающие функции, а выражение для W2 (E − E1 ) – быстро убывающая функция энергии. В результате вероятность P(E1 ) должна иметь максимум при некотором значении энергии E1 . Вместо функции P(E1 ) удобно рассматривать ее логарифм ln P(E1 ) = lnConst + lnW1 (E1 ) + lnW2 (E2 ). Экстремальное значение энергии E1 найдем из условия ∂ ln P 1 ∂P = = 0. ∂E1 P ∂E1 Это соотношение представляем в виде ∂ lnW1 (E1 ) ∂ lnW1 (E2 ) + (−1) = 0 ∂E1 ∂E2 или ∂ lnW1 (E1 ) ∂ lnW1 (E2 ) = = β, ∂E1 ∂E2 где β имеет размерность обратной энергии. Если считать, что внешние параметры системы фиксированы, как это и предполагалось, то условием равновесного состояния является равенство температур, т.е. T1 = T2 .(см. п. 1.6.). Тогда из термодинамического определения температуры T = ∂E ∂S , следует, что S1 = k lnW1 и S2 = k lnW2 . Соотношение S = k lnW будет справедливо для любого термодинамического объекта, находящегося в среде в равновесных условиях. 2.2 Принцип равной вероятности. Микроканоническое распределение Функции распределения для термодинамических систем определяют в энергетическом представлении, т. е. считают что в качестве случайной величины, представляющей систему, должна выступать ее энергия. Это определяется возможностью роли, которую играет энергия в физике и, в частности в термодинамике. Для классических систем 1 энергия выражается функцией Гамильтона H(X, a1 , a2 , . . .), где X – динамические переменные, ai – величины (параметры), 1 под классической понимаем систему, движение частиц которой представляется уравнением классической механики, например, в форме уравнений Гамильтона 14 определяющие соотношение системы с окружающей средой. Тогда, функцию распределения, представляющую состояние системы, следует записывать выражением f (X, a1 , a2 , . . .), где X – случайная величина. Если система рассматривается как квантовомеханический объект, то известной функции H(X, a1 , a2 , . . .) будет соответствовать целый набор собственных значений энергии системы En (a1 , a2 , . . .), где n – квантовые числа. Функция распределения, представляющая состояние квантовой системы определяется выражением ωn (a1 , a2 , . . .), где в данном случае в качестве случайной величины выступают квантовые числа n. Состояние системы с указанными значениями X либо n называются микросостоянием системы, а совокупность микросостояний – статистическим ансамблем (Гиббса). Переходим к установлению конкретного вида функции распределения. Будем считать, что классическая система изолирована, т. е. E = const. Функцию Гамильтона системы представим в виде H = H(X, a), где a – внешний параметр системы. Системе соответствует набор микросостояний W (a, E) – термодинамическая вероятность (см. п. 2.1). Постулируется, что все состояния, которым соответствует одно и тоже значение энергии, равновероятны. Используя это, точно записываем выражение для функции распределения системы в таких условиях f (X, a, E) = Cδ[H (X, a) − E] и Z f (X, a, E)dX = 1, где использовано представление δ – функции. Постоянную C находим из условия нормировки Z 1 C = [ δ[H(X, a) − E]dX]−1 = W (a, E) Распределение для квантовых систем при E = const записываем по аналогии с выше рассмотренными выражениями ω(a, E) = CδEn −E , ∑ ωn(a, E) = 1, n где используется представление символа Кронекера. Постоянная C находится из условия нормировки 1 C = [∑ δEn −E ]−1 , W (a, E) n где W (a, E) – число состояний для заданной энергии определяется квантовомеханической кратностью вырождения gn . 2.3 Каноническое распределение Теперь рассмотрим закрытую систему (см. п. 1.1), основное термодинамическое равенство для которой записывается в виде dE = T ds − Ada Будем учитывать, что энергия системы описывает случайные изменения. Однако температура T системы остается постоянной (T = const). Такая ситуация возможна 15 лишь в том случае, когда рассматриваемая система является малой подсистемой большого объекта (окружающей среды) находящегося в равновесных условиях. Считаем, что составная система рассматривается как изолированная. Тогда для нее используем микроканоническое распределение f (X1 , X2 , E) = Cδ[H(X1 ) + H(X2 ) + H(X1 , X2 ) − E] и Z f (X1 , X2 , E)dX1 dX2 = 1, где X1 , X2 – динамические переменные исследуемой системы и окружающей среды соответственно, H(X1 , X2 ) – энергия взаимодействия между системой и окружающей средой, E – полная энергия составной системы. Для упрощения записи в выражении для f (X1 , X2 , E) опущены зависимости от внешних параметров, которые в последующем будут учтены.Положим H(X1 ), H(X2 ) всегда гораздо большими, чем H(X1 , X2 ). Тогда для искомой функции распределения имеем Z f (X1 ) = f (X1 , X2 , E)dX2 . Учитывая постоянство температуры исследуемой системы , следует считать , что всегда выполняется H(X2 ) >> H(X1 ). В этом случае получаем Z f (X1 ) = C δ[H(X2 ) − E]dX2 = C Z δ[H(X2 ) − E]g(E2 )dE2 , где в последнем выражении перешли к интегрированию по энергии, g(E2 ) = dX2 dE2 – статистический вес для состояний окружающей среды. Теперь, учитывая рассматриваемую ситуацию 1 << E2 , полагаем полную энергию системы E равной энергии окружающей среды E2 , т.е. E2 = E = const. В этом случае величину g(2 ), которая представляет полное число состояний окружающей среды, следует рассматривать фактически как постоянную и для f (X1 ) получаем f (X1 ) = Cg(E − E1 ), где интеграл от δ функции равен единице, а энергия 2 = − 1 , где 1 – всегда существенно малая величина. Используем представление числа состояний системы через её энтропию (см.п.2.1).Тогда 1 g(E − E1 ) = exp[ S(E − E1 )]. k Осуществим разложение ∂S S(E − E1 ) = S(E) − E1 + ..., ∂E где ∂S ∂E = 1 1 = . T2 T1 16 Равенство температур 1 = 2 является следствием равновесного состояния. При этом следует считать, что внешние параметры для системы 1 и окружающей среды 2 , должны быть постоянными. Подстановка представленных выражений в функцию распределения даёт −E1 f (X1 ) = C1 exp . kT1 Эта формула справедлива для любого закрытого объекта, находящегося при постоянных и в достаточно большой макроскопической среде. Опуская индекс записываем в виде −H(X, a) f (X, a, T ) = Const exp , kT Z f (X, a, T )dX = 1, где случайная величина E1 заменена соответствующей функцией Гамильтона (X, a). Представим общую форму записи канонического распределения 1 −H(X, a) f (X, a, T ) = exp , z kT где Z z= −H(X, a) exp dX kT – статистический интеграл распределения. По аналогии записываем каноническое распределение для квантовых систем 1 −En ωn (a, T ) = exp , z kT где −En z = ∑ exp kT n – статистическая сумма распределения. 2.4 Обобщенное распределение Будем рассматривать открытую систему (см. п.1.1), основное термодинамическое равенство, для которой записывается в виде dE = T dS − Ada + µdN. В качестве случайных величин в данном случае выступают энергия системы и число частиц N. Функция Гамильтона будет определяться зависимостью H = H(X, a, N), где в данном случае X и N – случайные. Положим, что величины a, T, µ являются заданными. Получим выражение для функции распределения f (X, N, a, T, µ). Рассматриваем данную систему и окружающую среду как изолированную. Исходное микроканоническое распределение записываем в виде (индекс 1 – объект, индекс 2 – окружающая среда). f (X1 , X2 , N1 , N2 ) = σδ [H(X1 , N1 ) + H(X2 , N2 ) − E] , 17 где N = N1 +N2 , E = E1 +E2 , т.е. полагаем, что отсутствует энергия взаимодействия между системой и окружающей средой H(X1 , N1 , X2 , N2 ) = 0. Постоянная σ определяется условием нормировки: ∑∑ Z f (X1 , X2 , N1 , N2 )dXN1 dXN2 = 1. N1 N2 Искомую функцию распределения f (X1 , N1 ) представим через микроканоническое распределение f (X1 , N1 , X2 , N2 ): Z f (X1 , N1 ) = f (X1 , X2 , N1 , N2 )dXN2 Для рассматриваемой системы во всех случаях должно выполняться N1 << N2 и E1 << E2 . Это позволяет записать f (X1 , N1 ) = σ Z δ[H (X1 , N2 ) − E]dXN2 = σ Z δ[H (X2 , N2 ) − E]g (EN2 ) dEN2 , где g (EN2 ) = g (E2 , N2 ) = dXN2 dEN2 –статистический вес окружающей среды (для состояния 2 с числом частиц N2 ). Рассматривая g (EN2 ) как постоянную величину в данных условиях, записываем f (X1 , N1 ) = σ · g (E2 , N2 ) , где интеграл от дельта-функции полагаем равным единице. Выражение для статистического веса можно записать в виде (см. п. 2.1.) 1 g (E2 , N2 ) = exp S (E2 , N2 ) . k Энтропию S окружающей среды представляем выражением ∂S ∂S S2 (E − E1 , N − N1 ) = S (E, N) − E1 − N1 + ..., ∂E ∂N где ∂S ∂E a,N 1 = , T2 ∂S ∂N =− a,E µ2 . T2 В состоянии равновесия должно выполняться 2 = 1 и µ2 = µ1 . Используя эти представления и вводя новую постоянную, получим f (X1 , N1 ) = const · e µ1 N1 −EN 1 kT . Эта формула справедлива для любого открытого объекта, находящегося при постоянных , a, µ в достаточно большой макроскопической среде. Общая форма записи обобщенного распределения имеет вид f (X, N, a, T, µ) = 1 µN−H(X,N,a) kT e , Z 18 ∑ N Z f (X, N, a, T, µ)dXN = 1, где Z=∑ Z e µN−H(X,N,a) kT dXN N статистическая сумма обобщенного распределения. Для квантовых систем распределение ωn,N (a, T, µ) запишем по аналогии с классическим распределением ωn,N (a, T, µ) = 1 µN−En,N (a) e kT , Z где Z = ∑∑e ∑ ∑ ωn,N = 1, N n µN−En,N (a) kT N n .2 2.5 Связь квантовых и классических распределений При переходе от квантовых к классическим распределениям формально надо заменить суммирование ∑ . . . по возможным микросостояниям (n) квантовой системы интегрироR n вание ...dX по динамическим классическим переменным, например, по координатам и импульсам частиц системы dX = d~r1 d~p1 ...d~rs d~ps , где s – число степеней свободы системы. Однако, чтобы результаты расчетов в последнем случае для тех или иных величин были правильными, необходимо учесть два фактора. В квантовой механике для динамических переменных r и p существует соотношение (неопределенности) ∆ri · ∆pi ≈ h, где h – постоянная Планка. Тогда это соотношение будет определять число состояний для классической системы следующим образом: ∑ ... → Z ... n dX . hS Для системы из N свободных частиц S = 3N. 2. Это относится к случаю однотипных частиц. В квантовой механике формулируется принцип тождественности, или принцип неразличимости одинаковых частиц. Когда для квантовых систем проводим суммирование ∑ , то это означает, что проводится суммироn вание по всем физически различным состояниям системы ni , то перестановка частиц по "своим"состояниям не приводит к появлению дополнительных состояний системы. В классической физике одинаковых частиц все частицы являются различными. В простейшем случае эту ситуацию можно пояснить следующим образом. Допустим объект состоит из двух частиц: a и b. Частицы однотипны в том случае, что энергетический спектр состояний для них одинаков. Рассмотрим первую ситуацию. Частицы находятся в состояниях Ea,x1 и Eb,x2 , где xi = (~ri ,~pi ). Это микросостояние такой системы, энергия которой определяется Ea,x1 и Eb,x2 . Теперь переставим частицы по состояниям, т.е. будем иметь Ea,x1 и Eb,x2 . Это будет новое микросостояние системы, хотя физическое состояние - по энергии будет прежним. Вроде создается впечатление на таком простом примере, что перестановка частиц не влияет конечное значение энергии системы. Однако, это не так. Дело заключается в том, что все величины в статистической термодинамике 2 Вывод квантового распределения, например, в литературе: Климантович Ю.Л., Наука, М., стр.81 19 получают в результате усреднения с помощью функций распределения по всевозможR ным состояниям системы. Это означает, что при интегрировании dX по состояниям N частиц необходимо учитывать расположение различимых частиц по возможным для них состояниям. Это приведет к тому, что число таких интегралов будет N! (число перестановок из N частиц). Тогда с учетом принципа тождественности следует использовать условие перехода к результатам классической теории ∑ ... → n 1 N! dX .... h3N Z В результате переход от статистической суммы канонического распределения к интегралу происходит следующим образом Z = ∑e − En kT n 1 → N! Z e− H(X) kT dX . h3N Для статистических сумм обобщенного распределения получаем ∑∑e βN−En,N (a) kT N n 2.6 1 →∑ N N! Z e µN−H(X,N,a) kT dXN . h3N Дополнительные вопросы 1. Имеем частицу со спином равным 1/2 и магнитным моментом~µ0 во внешнем магнитном поле ~B. Пространственное положение частицы фиксировано. Магнитный момент частицы может быть направлен либо параллельно полю, либо антипараллельно. В первом случае энергия частицы равна −µ0 B, а во втором случае +µ0 B. Допустим объект состоит из N таких частиц, взаимодействие между которыми отсутствует. Определить полное число возможных состояний системы при N = 1, 2, 3, 4. Допустим при N = 4 энергия системы каким либо образом фиксирована и равна −2µ0 B. Установить число допустимых состояний в этом случае. Ответ: N = 1, W = 2; N = 4, W = 16W ; Wg (Em = −2µ0 B) = 4; энергетический спектр состоит из 5 уровней gm1 = 1, gm2 = 4 и т.д. 2. Для свободной классической частицы в объёме V полное число состояний W и 1 3 2 2 статистический вес g(E) = dW dE определяются выражениями W ∼ V E и g(E) ∼ V E , как следует из П.2.1. Получите выражение для W и g(E) системы из N свободных невзаимодействующих частиц. Согласны ли вы с утверждением, что для макроскопических тел число состояний "катастрофически"возрастает с увеличением энергии. 3. Частица массы m свободно перемещается в объёме V = L3 , где L – ребро куба. Собственные значения энергии частицы определяются выражением En = h̄2 π2 2 (n + n22 + n23 ), 2mL2 1 gni = 1, ni = 1, 2, . . ., n = (n1 , n2 , n3 ). Допустим такая модель находится в среде при = 300 К. В каких состояниях по квантовым числам n следует ожидать нахождение частицы. (для m взять массу электрона либо простейших молекул, L = 1 см). Полагая, что при больших n энергетический спектр частицы можно считать непрерывным, получите выражение для статистического веса такой системы Ответ: W = 18 ( 43 πn3 ), где n = 1 L 2 πh̄ (2mE) ; 1 g(E) ∼ V E 2 . 20 3 Статистическая термодинамика равновесных систем Функции распределения (Гиббса), рассмотренные в предыдущей части, открывают новые возможности для исследования свойств термодинамических систем. 3.1 Статистическое представление внутренней энергии Рассмотрим закрытую термодинамическую систему, состояние которой задаём функцией канонического распределения f (X, a, T ). Среднее значение энергии такой системы определяется выражением h i R Z H(X, a) exp − H(X,a) dX kT h i E(a, T ) = H (X, a) f (X, a, T ) dX = (56) R exp − H(X,a) dX kT Это выражение представляется в другой эквивалентной форме E(a, T ) = kT 2 где Z Z(a, T ) = d ln Z(a, T ), dT H(X, a) exp − dX kT Аналогичное представление E можно сделать для квантовых систем h i En (a) E (a) exp − ∑ n kT h i E(a, T ) = ∑ En (a)ωn (a, T ) = n En (a) n exp − ∑ kT (57) (58) (59) n или в аналогичном виде (58), где En (a) Z(a, T ) = ∑ exp − dX. kT n (60) 3.2 Статистическое представление числа частиц Рассмотрим открытую термодинамическую систему, состояние которой задаётся функцией обобщенного распределения. Среднее значение числа частиц определяется выражением h i R µN−H(X,N,a) dXn Z ∑ N exp − kT h i N (a, T, µ) = ∑ N f (X, N, a, T, µ) dXn = n R (61) µN−H(X,N,a) n dXn ∑ exp − kT n Эквивалентной формой этого представления является выражение N(a, T, µ) = kT где Z0 (a, T, µ) = ∑ n Z d ln Z0 (a, T, µ), dT µN − H(X, N, a) exp − dXn . kT 21 (62) (63) Аналогичные выражения определяются для квантовых систем h i µN−En,N (a) ∑ ∑ N exp − Z kT n N h i N (a, T, µ) = ∑ ∑ Nωn,N (X, N, a, T, µ) = µN−En,N (a) n N ∑ ∑ exp − kT (64) n N или в эквивалентной форме (62), где µN − En,N (a) Z0 (a, T, µ) = ∑ ∑ exp − . kT n N (65) 3.3 Свободная энергия (F) и омега-потенциал (Ω) Прежде чем обсудить метод расчета F и Ω, сделаем более общим представление энтропии системы в виде S = k lnW (см. п. 3.1). Число доступных состояний системы W определяется средним значением энергии E системы. В этом случае функция распределения f [H (X)] существенна для микросостояний X, у которых H (X) = E. Запишем условие нормировки для f [H (X)] в виде h iZ h i f H (X) dX = 1 f H (X) W = 1. (66) Тогда получаем h i S = k lnW = −k ln f H (X) . (67) Конструкция функций распределения (Гиббса) позволяет переносить усреднение с аргумента H(X) на функциональное выражение ln f [H (X)]. В этом случае приходим выражению Z S = −kln f [H (X)] = −k ln f (x, a, T ) · f (x, a, T )dx. (68) Аналогичные рассуждения можно провести для квантовых систем, что приводит к выражению S = −k ∑ ln ωn · ωn . (69) n Теперь перейдем к рассмотрению термодинамических потенциалов. Используем выражение для энтропии в виде (67), где H (X) f [H (X)] = C exp − , (70) kT где C – постоянная. Усреднение приводит к результату " # H(X) E S = −k lnC − =, −k lnC − kT kT (71) Будем отождествлять среднюю энергию E с энергией термодинамической . Используем термодинамическое определение свободной энергии (??). Тогда из последнего выражения для энтропии (71) получаем F(a, T ) = −kT ln Z(a, T ), 22 (72) где Z(a, T ) – статистический интеграл канонического распределения. Аналогичное выражение для F справедливо и для квантовых систем, если для Z принимать статистическую сумму канонического распределения. Такое представление свободной энергии F позволяет записать каноническое распределение в виде ωn (a, T ) = exp [F(a, T ) − En (a)kT ] . f (x, a, T ) = exp [F(a, T ) − H(x, a)kT ] , (73) Запишем обобщенное распределение в виде f (X, N) = C exp [µN − H(X, N)kT ] , где = 1/Z0 . Подстановка f (X, N) в выражение (67) приводит к результату µN − E S = −k lnC − . kT (74) (75) Отождествляем E и N c термодинамическими значениями E и N. Используем определение для омега-потенциала Ω = E −T S −µN (см. п.1.5). Тогда из выражения для энтропии S получаем Ω(a, T, µ) = −kT ln Z0 (a, T, µ). (76) Это выражение справедливо и для квантовых систем. Такое представление омегапотенциала Ω позволяет представить обобщенное распределение в виде f (x, N, a, T, µ) = exp [F(a, T ) − H(x, a)kT ] , ωn (a, T ) = exp [F(a, T ) − En (a)kT ] . (77) В заключении дополним еще одним представлением для энтропии S системы, наряду с рассмотренными выражениями в начале параграфа. По определению ST = E − F,где ∂ F = −kT ln Z и E = kT 2 ∂T ln Z. После подстановки получаем S=k ∂ (T ln Z) . ∂T (78) 3.4 Методы статистической термодинамики Феноменологическая термодинамика (1) позволяет исследовать все макроскопические величины системы, при условии, что для нее известны уравнения состояния либо термодинамические потенциалы. Статистическая термодинамика предлагает метод расчета уравнений состояний системы. Калорическое уравнение состояния получается из выражения ∂ E(a, T ) = kT 2 ln Z(a, T ). (79) ∂T Термическое уравнение состояния определяют выражением ∂F(a, T ) , (80) ∂a где F(a, T ) = −kT ln Z(a, T ). При этом, получаемые зависимости E = E(a, T ) и A = A(a, T ) представляется через стандартную пару независимых переменных (a, T ), что находится в соответствии с первым (нулевым) началом термодинамики (см. п. 1.2). Выражения F(a, T ) = −kT ln Z(a, T ) и Ω(a, T, µ) = −kT ln Z0 (a, T, µ) – определяют термодинамические потенциалы F и Ω в своих естественных переменных. Статистические выражения получают и для остальных термодинамических потенциалов. Однако, в этих случаях зависимости носят достаточно сложный характер (см. Дополнение, вопрос 2.??). Отметим, что все величины термодинамики в принципе можно представить через Z либо Z0 . Поэтому методику расчета величин в статистической термодинамике называют методом статистических сумм (интегралов). A(a, T ) = − 23 3.5 Статистическое обоснование начал феноменологической термодинамики Используем условие нормировки для канонического распределения Z Z F(a, T ) H(x, a) f (x)dx = exp exp − dx = 1. kT kT (81) Продифференцируем это равенство, считая, что T и a испытали малые изменения Z Z F F H E H 1 ∂H 1 exp d exp − dx exp exp − da + Hd dx = 1. kT kT kT kT kT kT ∂a kT (82) Если учитывать условие нормировки, получаем 1 F 1 ∂H d − da − +Hd = 0, (83) kT kT ∂a kT где H = E – энергия системы, ∂H da = dW ∂a – термодинамическая работа. Таким образом имеем F 1 Td − T Ed = dW. T T Используем представление E −F F 1 Td − T Ed = dE − T d = dE − T dS, T T T (84) (85) (86) где (E − F)/T = S -энтропия. Окончательно получаем dE = T dS + dW – основное термодинамическое равенство для закрытой системы (см. 2). Энтропия квантовой системы может быть представлена выражением (78), где Z = En ∑n gn exp − kT . Рассмотрим случай, когда kT << E1 − E0 , где E1 –первый возбужденE0 ный уровень энергии для системы. Тогда получаем Z = Z0 n = g0 exp − kT , а для энтропии приходим к результату limS = S0 = k ln g0 = const, что соответствует третьему началу термодинамики (см.5). 3.6 Дополнительные вопросы 1. Система состоит из почти не взаимодействующих подсистем A, B, C,... Показать, что при определенных допущениях статистическая сумма канонического распределения представляется в виде Z = ZA ZB ZC , где ZA , ZB , ZC ,... – статистические суммы подсистем A, B, C,... 2. Представить термодинамические потенциалы Φ-Гиббса и -энтальпии через статистическую сумму ( интеграл ) канонического распределения. 3. Допустим системе соответствуют только два энергетических уровня E1 и E2 (E2 > E1 ) c кратностью вырождения g1 и g2 соответственно. В первом случае системе сообщают некоторое количество теплоты ∆Q так, что ее температура изменилась от T0 до T1 . 24 Определить ∆Q в таком процессе. Во втором случае система адиабатически изолирована (∆Q = 0 ) и над ней совершается работа W , так что уровень E2 смещается и принимает значение E20 = E2 + ∆E. Определить W для такого процесса. 4. Статистический вес g(E) для некоторой системы имеет вид а) g(E) ∼ exp(−αE), б) g(E) ∼ exp(+αE), где α - положительная постоянная. Возможно ли рассчитать свойства таких систем методами равновесной статистической термодинамики. 4 Идеальные ТД системы в равновесном состоянии. Статистика Больцмана Применим представления равновесной статистической ТД к объектам с конкретными типами функций Гамильтона H. В данном случае систему определяем функцией вида N H (X) = ∑ H (xi ), i=1 где X = (x1 , x2 , ..., xN ) – совокупность всех динамических переменных, определяющих состояние систем из N – частиц, xi = (~ri ,~pi ) – динамические переменных, соответствующие состоянию отдельной частицы системы. Функцию Гамильтона H(Xi ) на отдельную частицу представляется в виде H (xi ) = Ek (~pi ) + E (~ri ) , где Ek (~p) – полная кинетическая энергия теплового движения частицы, En (~r) – полная потенциальная энергия частицы во внешнем поле. Системы с таким видом функций H (X) называются идеальными системами. Как будет показано для таких систем методы статистической ТД получают дополнительное развитие. Будем считать, в данной части, что система находится в таких условиях, когда индивидуальные свойства частиц, связанные с наличием у них спина, не проявляются. Это случай классической статистики( Больцмана ). 4.1 Классическое распределение Максвелла-Больцмана Для определенности рассмотрим систему, состоящую из N однотипных "бесструктурных"частиц, находящихся в термодинамическом равновесии при температуре T в V . Состояния частиц определяются классически. Будем считать, что рассматриваемая модель относится к закрытым ТД системам. Тогда ее состояние представляется функцией канонического классического распределения (см. п. 2.3.). 1 H(X) f (X) = e− kT , z где z = e− R H(X) kT dX; R f (X) dX = 1 В данном случае имеем N ∑ H(xi ) 1 − kT1 i=1 f (X) = e , z где H (xi ) = Px2i +Py2i +Pz2i 2m + E (xi , yi , zi ) = ~p2i 2m + E (~ri ), m – масса частицы. 25 Если точно указана аналитическая зависимость xxx и обозначены интервалы значений и , то следует считать, что все ТД свойства рассматриваемой модели могут быть установлены. Для иллюстрации запишем ряд очевидных выражений Z H (X) · f (X) dX E = H̄ (X) = – полная энергия системы, Z H X~p · f (X) dX Ek = H̄ X~p = – полная кинетическая энергия системы, Z H (X~r ) · f (X) dX E = H̄ (X~r ) = – полная потенциальная энергия системы, ~p2i = 2m ~p2i · f (X) dX 2m Z – кинетическая энергия теплового движения на одну частицу, Z E (~ri ) · f (X) dX Ē (~ri ) = – потенциальная энергия частицы во внешнем поле. В то же время для идеальных систем математическое представление исследуемых выражений можно существенно упростить. Это связано с тем, что для идеальных систем состояние любой частицы статистически не зависит от состояния других частиц. Тогда состояние каждой частицы определяется с помощью "индивидуальной"функции распределения f (xi ). Из условия идеальной системы следует N f (X) = ∏ f (xi ). i=1 Кроме того между функциями распределения должно существовать соответствие Z f (xi ) = f (X) dXN−1 , Где интегрирование ведется по координатам и импульсам всех частиц системы. Любые из приведенных выражений позволяют установить конструкцию функции распределения, f (xi ). Остановимся на втором представлении. R − kT1 ∑ H(xi ) f (xi ) = e R i 1 − kT e dx1 dx2 ...dxN−1 ∑ H(xi ) i dx1 dx2 ...dxN Откуда получаем f (x) = R e− e− 26 H(x) kT H(x) kT , dx где индекс "i"опущен, т.к. это представление справедливо для любой частицы системы. Назовем f (x) – одночастичной функцией распределения для классических функций распределения для классических идеальных систем (распределение МаксвеллаБольцмана). Запишем ее в виде f (x) = 1 e zMB 1 − kT Z zMB = 1 − kT e 2 2 p2 x +py +pz +E(x,y,z) 2m 2 2 p2 x +py +pz +E(x,y,z) 2m – статистический интеграл распределения Максвелла-Больцмана. Определим более простые одночастичные функций распределения: 1.Распределение Максвелла Z f (px, py, pz ) = f (x) dxdydz или 1 − p2x +p2y +p2z f (px, py, pz ) = e 2mkT , zM = zM 2.Одномерное Распределение Максвелла Z e− 2 2 p2 x +py +pz 2mkT d px d py d pz . Z f (px ) = или f (px ) = 1 f (px , py , pz ) d py d pz , p2 x e− 2mkT , 1 zM1 = (2πmkT ) 2 , z M1 где при интегрировании zM1 использовались пределы интегрирования −∞ < px < +∞, что приводит выражение к табличному интегралу. 4.1.1 Приложение 1. При использовании для расчетов распределения в виде f (~p) либо f (~v) применяются интегралы вида Z∞ In = 2 e−αx xn dx, 0 где α > 0. Для n = 0 √ I0 = π −1 α 2. 2 Для n = 1 1 I1 = α−1 . 2 Для остальных интегралов при n ≥ 2 (n – целое) результат получают через I0 и I1 последовательным интегрированием по частям, что дает n−1 In = In−2 2α 27 Например, √ I0 π −3 I2 = = α 2 2α 4 2.Если распределение Максвелла представлено через энергию, т.е. f (E), то для расчетов применяются интегралы вида Γ (z) = Z∞ xz−1 e−x dx 0 – интегралы типа гамма-функций, где x – вещественная переменная, z – принимает целые и полуцелые значения. Γ (1) = Z∞ e−x dx = 1 0 , Z∞ √ 1 1 Γ = x− 2 e−x dx = π 2 0 . Остальные интегралы определяются из рекуррентного соотношения Γ (z + 1) = zΓ (z) . 3. Классическое распределение Больцмана Z f (x, y, z) = или f (x) d px d py d pz 1 E(x,y,z) f (x, y, z) = e− kT z E(x,y,z) , где z = e− kT dxdydz. В частном случае E (x, y, z) = 0 имеем R Z e− z= E(x,y,z) kT dxdydz Окончательно распределение Максвелла-Больцмана записывается в виде 1 − kT f (~r,~p) = e 2 2 p2 x +py +pz +E(x,y,z) 2m 3 (2πmkT ) 2 e− R E(x,y,z) kT . dxdydz Здесь следует сделать замечание. Как следует из определения, распределение Максвелла-Больцмана справедливо только для идеальных систем. Распределение Максвелла применимо к любым классическим объектам. Действительно, функция Гамильтона представляется в виде H (X) = ∑ i ~p2i + E (~r1 ,~r2 , ....) , 2m 28 где E – определяет полную потенциальную энергию частиц (взаимодействия и во внешнем поле). Каноническое распределение можно представить в виде f (X) = f X~p · f (X~r ) , что свидетельствует о статистической независимости распределение частиц по координатам X~r и импульсам X~p . При этом выполняется f X~g = ∏ f (~pi ), i=1 где f (~pi ) – распределение Максвелла. Преимущество использования функции распределение Максвелла-Больцмана продемонстрируем на расчетах, приведенных в следующем параграфе. 4.2 Термодинамические свойства классического газа "бесструктурных"частиц Рассмотрим систему, состоящую из N однотипных свободных частиц массы m занимающих объем V при температуре T . Состояние системы задаем одночастичной функцией распределение Максвелла-Больцмана. Определим ряд величин, относящихся непосредственно к частицам: 1. Среднее значение проекции импульса (px ) теплового движения частицы Z+∞ px · f (px ) d px = p̄x = Z+∞ 1 1 p2 x px e− 2mkT d px = 0, (2πmkT ) 2 −∞ −∞ где учтено, что интеграл с симметричными пределами от нечетной подынтегральной функции равен 0. 2. Среднее значение импульса Что дает в соответствии с П.1 3. Среднее значение величины импульса ~p Z∞ p̄ = p · F (p) d p, F (p) = f (~p) · 4πp2 0 или Z∞ 4π p̄ = (2πmkT ) 3 2 2 p 3 − 2mkT p e 0 dp = 8mkT π 1 2 , где использовано представление табличного интеграла (Приложение). 4. Среднее значение величины скорости ~v Z∞ v̄ = F (v) = f (~v) · 4πv2 v · F (v) · dv, o и f (~v) = 2 2 m 3 m(v2 x +vy +vz ) 2 · e− 2mkT 2πkT 29 Окончательно получаем v̄ = 8kT πm 1 2 . 5. Наиболее вероятное значение величины импульса dF (p) = 0, dp 1 pB = (2mkT ) 2 . 6. Среднее значение кинетической энергии теплового движения частицы, где Ek = 2 или Ek = mv2 . Тогда получаем, например, используя первое представление: Ek = p2 2m Z∞ 2 p 3 F (p) d p = kT, 2m 2 0 где использовано представление табличного интеграла (Приложение 4.1.1). Отметим, что определение среднего Ēk можно получить по собственной функции распределения f (Ek ): Z∞ Ek = Ek f (Ek ) dEk , 0 где E 2 1 − kT f (Ek ) = √ e E 1/2 3/2 π (kT ) выражение для которой следует из распределения F (p) или F (v). Значение табличного интеграла для E k приведено в Приложении (п. 4.1). 7. Среднее значение потенциальной энергии частицы в однородном гравитационном поле. Имеем E (z) = mgz, где g = const. Тогда получаем: Z∞ E= mgz ∗ f (z)dz, 0 где f (z) – одномерная функция распределения Больцмана. В данном случае mgz f (z) = R∞ e− kT mgz e− kT dz . 0 Непосредственное интегрирование в выражении для En приводит к результату En = kT . Теперь обсудим некоторые термодинамические свойства рассматриваемой системы. Как следует из общих представлений статистической термодинамики (Часть III), для этого достаточно определить статистический интеграл системы. В нашем случае имеем: N Z z= 1 − kT ∑ H(xi ) e i=1 dx1 dx2 ...dxN или z = zN 30 и Z z= e− H(x) kT dx , где z – статистический интеграл распределения Максвелла-Больцмана. Таким образом, получен важный результат. Всю информацию о свойствах системы можно получить через статистический интеграл одночастичной функции распределения. При дальнейших расчетах в выражении для Z учтем принцип неразличимости тождественных частиц и соотношение неопределенности (Часть III). Это приводит к представлению: 1 z = zN , N! где Z − H(x) e kT dx, z= h3 h – постоянная Планка. Если положить, что E (~r) = 0, тогда получаем: 3N (2πmkT ) 2 V N z= N!h3N Определим термодинамический потенциал F - свободная энергия: V 3N 3 F = −kT lnz = −kT Nln + ln (2πmkT ) − Nlnh + N , N 2 где использовано lnN! = NlnN − N (формула Стирлинга). Определим термическое уравнение состояния системы: ∂F NKT p=− = . ∂V T,N V Калорическое уравнение состояния можно получить из выражений E = F + T S, где ∂F S=− ∂T . V,N Это уравнение можно получить и самостоятельно: E = kT 2 ∂ 3 lnz = N kT. ∂T 2 Химический потенциал системы: ∂F Nh3 µ= = kT ln . ∂N V,T (2πmkT )3/2 V Термодинамическая вероятность системы (число состояний системы): Z W= dX 1 = 3N h N! N!h3N 31 Z dx1 dx2 ...dxN или Z 1 W= N! dx h3 N = 1 N W N! i Определим выражение для Wi , которое соответствует термодинамической вероятности на одну частицу: Z Wi = dx 1 = 3 3 h h Z или Wi = где 2 dxdydz ∗ 4πp d p = 4 3 3V p h3 4πV (2mE)3/2 , 3h3 p2 . 2m Тогда статистический вес на одну частицу представляется в виде: √ 4 2πm3/2V 1/2 g(E) = E h3 E= Обсуждение термодинамических свойств этой системы будет продолжено в последующих параграфах. 4.3 Одночастичное распределение Больцмана для систем с дискретным спектром состояний В данном случае рассматриваются идеальные системы в предположении, что информация о состояниях получена методами квантовой механики. Для идеальной системы из N частиц полная энергия En равна сумме энергий Eni отдельных частиц: N En = ∑ Emi , i=1 где mi - квантовые числа, определяющие состояние отдельной i-й частицы, n - квантовые числа, определяющие состояние системы. Очевидно, что в случае идеальной системы ее состояние будет определяться набором квантовых чисел, соответствующих отдельным частицам системы, т.е. n = (m1 , m2 , ..., mN ). Будем считать, что рассматриваемый объект относится к термодинамическим системам закрытого типа. Тогда статистическое состояние объекта определяется функцией канонического распределения: 1 EN ωn = e− kT , z z = ∑ e− kT . En n В то же время, вследствие идеальности объекта может быть определена одночастичная функция распределения ωmi . 32 Получим конкретный вид ωmi путем обобщения одночастичной классической функ−r , → −p ). Для функции f (→ −r , → −p ) имеем (см. ции распределения Максвелла-Больцмана f (→ п.1): → −→ − H( r , p ) − → − → − kT f ( r , p ) = C1 e где C1 - постоянная. Тогда по аналогии для функции ωm получаем: Em ωm = C2 e− kT где C2 - постоянная и индекс при m опущен, т.к. считаем, что распределение справедливо для любой частицы. Постоянную C2 распределения устанавливаем из условия нормировки: ∑ ωm = 1, m где 1 C2 = Em ∑ e− kT . m Окончательно записываем в виде: 1 Em ωm = e− kT z и z = ∑ e− kT . Em m Такое распределение ωm называют одночастичным распределением Больцмана, а z статистической суммой распределения Больцмана. По тексту предполагалось, что это распределение получено для квантовых идеальных систем. Однако, при обсуждении использован только тот факт, что квантовые системы должны иметь дискретный энергетический спектр и состояние задается квантовыми числами. В то же время, не затрагивался вопрос об индивидуальных свойствах частиц, связанных с наличием у них спина. В связи с этим, представленное распределение Больцмана следует рассматривать как распределение для идеальных систем с дискретным спектром состояний, у которых дискретность связана с квантовыми явлениями. Точное одночастичное квантовое распределение (распределение Ферми, распределение Бозе) будет получено в части 5. Распределение Больцмана является частным случаем этих распределений. Оно позволяет достаточно точно проводить расчеты свойств термодинамических систем, для которых детальное квантовомеханическое представление можно не учитывать. Установим связь между статистической суммой канонического распределения Z и статистической суммой одночастичного распределения z. Для конкретной конфигурации частиц записываем: En 1 z = ∑ e− kT = ∑ e− kT [Em1 +Em2 +...] n m1 ,m2 ,... − EkTm или z = zN , где Z = ∑ e , если считать частицы однотипными. С учетом возможных m перестановок частиц по состояниям, окончательно в этом случае получаем: z= 1 N z , N! 33 где N - полное число частиц в системе. В статистической термодинамике информацию о свойствах системы получают через статистическую сумму (интеграл) системы. Очевидно, что для идеальных систем с дискретным спектром состояний свойства системы могут быть установлены через статистическую сумму (интеграл) одночастичного распределения. Это позволяет считать, что состояние идеальной системы определяется одночастичным распределением. 4.4 Термодинамические свойства квантового больцмановского газа "бесструктурных"частиц Рассмотрим объект из N однотипных "бесструктурных"частиц, занимающих объём V при температуре T . Собственные значения энергии отдельной частицы могут быть представлены выражением: Em1 ,m2 ,m3 h2 2 2 2 = m + m + m , 1 2 3 8m0 L2 где mi = 1, 2, 3, ... – квантовые числа, и кратность вырождения gEmi = 1. Рассмотрим статистическую сумму, соответствующую отдельной частице: h2 2 2 2 z = ∑ ∑ ∑ exp − m1 + m2 + m3 8m0 L2 kT m1 m2 m3 или " ∞ h2 z = ∑ exp − m2 2 kT 8m L 0 m=1 # 3 . Таким образом, в принципе вопрос о термодинамических свойствах такой модели решен, т.к. статистическая сумма системы равна: " #3N ∞ 1 h2 z= . ∑ exp − 8m0L2kT m2 N! m=1 По установленной Z однозначно определяются калорические и термические величины. Единственное, что остается в данном случае - это приведение z к более простому аналитическому виду3 . Покажем, что для реальных ситуаций нет необходимости в точном определении статистической суммы. Действительно, определим характеристическую температуру поступательного (теплового) движения: h2 1 T= . 8m0 L2 k Допустим L = 1 см. и m соответствует массе электрона. Тогда Tx = 10−10 K и множи2 тель 8mh L2 ∼ = 10−27 эрг ∼ = 10−15 эВ. Для реальных температур T перераспределение энер0 гии между объектом и окружающей средой происходит "на уровне"порядка kT (считаем,что окружающая среда рассматривается как классическая система). Тогда собственные значения Em достаточно существенны. Считая Em ∼ kT , полагаем, например, T ≈ 3 Такой метод предлагается, например, в литературе: Ю.Б. Румер, М.Ш. Рывкин “Термодинамика, ста- тистическая физика, кинетика”, Наука, М., 1977. 34 300 K, имеем для Em ≈ 0.025 эВ. Тогда во всех реальных случаях частица будет находится в состояниях с очень большим квантовым числом m по всем степеням свободы (m1 , m2 , m3 ). В этом случае квантованием энергии для поступательного движения частиц в термодинамических системах несущественно и, следовательно, энергетический спектр будет квазинепрерывный. Состояние каждой частицы можно задавать классически (~r,~p) , а энергию системы представлять функцией Гамильтона H(~r,~p), как это делается в §2, где рассматривался классический идеальный газ. Соответственно, распределение частиц по состояниям будет определяться функцией распределения Максвелла – Больцмана, с помощью которой могут быть установлены все термодинамические свойства такой системы. В заключение покажем, что статистический вес g(E) при больших m соответствует классическому выражению. При m1 , m2 , m3 – больших, выражение h2 Em = m2 2 8mL можно рассматривать как непрерывную функцию числа m. Число состояний dW частицы в интервале квантовых чисел (m, m + dm) равно dW = 4πm2 dm , 8 или после подстановки m2 получаем √ 3 4 2πm 2 V 1 dW = g(E)dE = E 2 dE h3 Этот результат точно соответствует расчетам статистического веса классической частицы (см. §2). 4.5 Статистические суммы системы квантовых осцилляторов и квантовых ротаторов Рассмотрим две модели термодинамических систем, свойства которых расчитаем, используя распределение Больцмана. Будем считать, что система состоит из N одномерных гармонических квантовых осцилляторов, совершающих колебания с частотой ω. Систему рассматриваем как идеальную. Это предполагает, что колебательный процесс для каждого осциллятора не связан с остальными. В тоже время вся система находится в термодинамическом равновесии при температуре T . Определим статистическую сумму отдельного осциллятора. Как известно, собственные значения энергии гармонического осциллятора равны 1 Em = h̄ω(m + ); 2 gEm = 1, где m = 0, 1, 2, 3, . . . – квантовые числа, gm – кратность вырождения. Статистическая сумма будет определяться выражением ∞ z= ∞ h̄ω h̄ω h̄ω 1 − 2kT (m + exp[− )] = e e− kT m ∑ ∑ kT 2 m=0 m=0 35 Рассматривая это выражение, как геометрическую прогрессию, запишем z в виде её суммы h̄ω e− 2kT z= h̄ω . 1 − e− kT Это выражение можно переписать в более общем виде 1 z= e h̄ω 2kT h̄ω − 2kT −e h̄ω = [2sh( )]−1 kT Статистическая сумма системы осцилляторов определяется выражением z= h̄ω 1 [2sh( )]−1 N! kT h̄ω h̄ω Рассмотрим частный случай когда kT >> h̄ω. Осуществим разложение e− 2kT и e− kT в степенной ряд. Тогда получаем z= h̄ω 1 − 2kT +... 1 − (1 − h̄ω kT + . . .) = (1 − h̄ω kT kT ) ≈ . 2kT h̄ω h̄ω Для выяснения полученного результата рассмотрим осциллятор как классическую модель. Определим статистический интеграл осциллятора Z+∞ z= exp[− −∞ 1 p2x αx2 d px dx ( + )] . kT 2m 2 h После интегрирования получаем z= 2πkT m 12 . h α 1 kT 2 Учтём υ1 = 2π m , где υ – частота, ω = 2πυ. Тогда получаем z = h̄ω , что соответα ствует квантовой статистической сумме при kT >> h̄ω. Таким образом, если определить характеристическую температуру осциллятора T = h̄ω k , то при T >> T расчёт статических свойств осциллятора можно проводить в классическом представлении. Рассмотрим теперь систему из N однотипных квантовых ротаторов, имеющих две степени свободы. Собственные значения энергии отдельной частицы равны Em = h̄2 m(m + 1) 2J gEm = 2m + 1, где J – момент энерции вращающейся частицы, m – квантовое число (m = 0, 1, 2, . . . ). Статистическая сумма ротатора определяется выражением ∞ z= ∑ (2m + 1) exp[− ?=0 1 h̄2 m(m + 1) ( )]. kT 2J Для получения статистической суммы системы, полученное z просто возводим в степень N и делим на N! . 36 2 h̄ Рассмотрим выражение для z для случая T >> Tx = 2Jk – характеристическая температура ротатора. В таких условиях сумму можно заменить интегралом Z∞ Z= TX m(m + 1)]dm. T (2m + 1) exp[− 0 Полагая m(m + 1) = x, получаем Z∞ z= 2JkT T = 2 . Tx h̄ Tx e− T x dx = 0 Рассмотрим эту модель в классическом представлении. Энергия вращательного движения определяется формулой P2 Pθ2 + sinϕ2 θ H(θ, ϕ, Pθ , Pϕ ) = 2J , где Θ и φ – полярный и азимутальный углы, PΘ и Pφ – обобщённые импульсы. Классический статистический интеграл имеет вид 1 z= 2 h Zπ dθ 0 Z+∞ Z2π dϕ dPθ −∞ 0 P2 Z+∞ dPϕ exp[− Pθ2 + sinϕ2 θ −∞ 2J ]; Интегрируя сначала по φ, PΘ , Pφ и затем по Θ, получим z= 8π2 JkT 2JkT = 2 , 2 h h̄ что соответствует результату, получаемому для статистической суммы. Таким образом, и для этой модели можно утверждать, что при T >> T расчёт статических свойств можно проводить в классическом представлении. Приведённые примеры позволяют сделать вывод, что если объект находится при некоторой температуре T , для которой kT >> Ei − E j , где Ei , E j – собственные значения энергии. Разность для термодинамических свойств можно использовать классическое распределение. 4.6 Распределение Больцмана для чисел заполнения Запишем выражения, представляющее распределение частиц системы по состояниям. Они получаются, если задано полное число частиц в системе и определены одночастичные функции распределения. Рассмотрим систему с дискретным спектром состояний. Для неё известно распределение 1 Em ωm = e− kT . z Тогда среднее число частиц, сопутствующее m. состоянию равно Nm = N − Em e kT , z? 37 ∑ Nm = N. m Аналогично для классического объекта. Имеем 1 H(x) f (x) = e− kT . z Отсюда среднее N(x) определяется выражением N − H(x) N(x) = e kT , z?? Z N(x)dx = N. Из последнего выражения N(x) легко получаются выражения для N(Px , Py , Pz ) и N(x, y, z) R N(Px , Py , Pz ) = N(x)dxdydz = R N(x, y, z) = N(x)dPx dPy dPz = H(P ,P ,Pz ) N − xkTy zm e N − H(x,y,z) kT . z? e , Такое представление функций распределений называют числами заполнения. Запишем в другой эквивалентной форме выражения для чисел заполнения. Для этого получим в общем виде выражения для химического потенциала µ идеальных систем. По определению имеем ∂F 1 zN µ= , F = −kT ln z = −kT ln . ∂N T,a N! N! Отсюда получаем N , z? где z – статистическая сумма (интеграл) одночастичного распределения. В связи с этим распределение Nm , N(x) представляются в виде µ = kT ln Nm = e µ−Em kT , N(x) = e µ−H(x) kT , где условия нормировки для Nm и N(x) прежние. При заданном N условия нормировки в этом случае можно рассматривать как выражения, представляющие (в неявном виде) химический потенциал системы. 4.7 Больцмановский газ с двумя энергетическими уровнями Применили выше полученные распределения к идеализированной модели, состоящей из N невзаимодействующих частиц, имеющих только два энергетических уровня E1 и E2 (E1 < E2 , E1 =0) с кратностью вырождения g1 и g2 соответственно. Для частиц системы при температуре имеем N N(E1 ) = g1 , Z N E2 N(E2 ) = g2 e− KT , Z E2 где Z = g1 + g2 e− KT . Химический потенциал µ определяем из условий нормировки N(E1 ) + N(E2 ) = N. Тогда получим µ−E2 µ g1 e KT + g2 e KT = N. 38 где µ = KT ln N E2 g1 + g2 e− KT энергия системы определяется выражением E = N(E1 ) · E1 + N(E2 ) · E2 В нашем случае получаем E = N(E2 ) · E2 = NE2 g2 E2 E2 g1 + g2 e− RN · e− KT . Теплоёмкость системы V находим по формуле V =( E2 2 E2 Ng1 g2 ( KT ) ∂E e− KT )V = E2 ∂T (g1 + g2 e− KT )2 Отсюда следует, что CV → 0 при T → 0 и при T → ∞, что объясняется идеализацией рассматриваемой модели. 4.8 Термодинамические свойства молекулярного газа. Квантовоклассическое распределение Больцмана Сейчас мы учтем, что для реальных объектов, состоящих из частиц, необходимо учитывать внутренние степени свободы. В распределении ωm = 1 − Em e KT Z? индекс m – обозначает полный набор квантовых чисел, характеризующих состояние частицы и учитывающих как внутреннее состояние, так и движение частицы как целого. Будем считать, что поступательное движение описывается классически, а внутреннее – квантовомеханически. Тогда выражение для энергии можно представить в виде → −2 p −r ) + E , + E(→ Em = i 2m где два первых слагаемых определяют кинетическую и потенциальную энергии частицы −r , → −p ), а E – внутренняя энергия частицы в состоянии i. в классическом состоянии (→ i Распределение Больцмана ωm в этом случае записывается в виде → −2 p → − −r , → −p ) = 1 exp[− µ − 2m + E( r ) + Ei ], ωi (→ Z kT где Z Z= . → −2 p → − −r d → −p d→ 2m + E( r ) + Ei exp[− ] h3 ∑ kT i Распределение для чисел заполнения Nm приобретают вид → −2 p −p ) − E µ − 2m − E(→ i → − → − Ni ( r , p ) = exp[− ], kT 39 где → −2 p −r ) − E → − → − µ − 2m − E(→ drdp i exp =N ∑ 3 h kT i Z . Таким образом, распределение Больцмана одновременно определяет распределение по координатам и импульсам – по классическим состояниям и по квантовым состояниям частиц (квантово-классическое распределение Больцмана). −r , → −p ) следует и частные распреОчевидно, что из этого общего представления ωi (→ деления, рассмотренные ранее. Действительно, чтобы перейти, например, к распределе−r , → −p ) просуммировать по всем кваннию Максвелла-Больцмана надо выражение ωi (→ товым состояниям частицы → −2 p → − 2m + E ( r ) −r , → −p ) = 1 ω (→ −r , → −p ) = 1 exp f (→ − , i h3 ∑ Z kT i где −r ) 3/ Z E (→ −r . Z = (2πmkT ) 2 exp − d→ kT Рассмотрим термодинамические свойства идеальной молекулярной газовой среды. Запишем выражение для статистической суммы системы Z= 1 N Z , N! где N – полное число молекул массы m в объёме V при температуре . Считаем, что −r ) = 0). внешнее поле отсутствует (E (→ Для Z имеем 3 E (2πmkT ) 2 − EkTm − kTi Z = ∑e = e . ∑ h3 m i Для молекулы надо учитывать, что её энергия Ei определяется колебательными и вращательными степенями свободы и электронным возбуждением. Будем считать Ei = Ei1 + Ei2 + Ei3 , где Ei1 ,Ei2 ,Ei3 – колебательная, вращательная энергия и энергия электронного возбуждения соответственно. Тогда для Z получим 3 (2πmkT ) 2 V Z1 · Z2 · Z3 , Z= h3 Ei1 Ei3 Ei2 где Z1 = ∑ e− kT , Z2 = ∑ e− kT , Z3 = ∑ e− kT – статистические суммы соответствующих i i i степеней свободы. Таким образом, для Z системы имеем 3N (2πmkT ) 2 V N N N N Z1 · Z2 · Z3 . Z= N!h3N 40 Обсудим некоторые термодинамические свойства молекулярной среды. Выражение для свободной энергии F системы в предположении отсутствия внешнего поля имеет вид V 3N 3 F = −kT ln Z = −kT N ln + ln (2πmkT ) − N ln h + N −NkT ln Z1 −NkT ln Z2 −NkT ln Z3 , N 2 где выражение в квадратных скобках определяет свободную энергию поступательного теплового движения молекул (см. п.4.2). Отсюда получаем, что термическое уравнение состояния системы представляется формулой ∂F NkT P=− = , ∂V T,N V где учтено, что в выражении для F все слагаемые, кроме первого, от объёма не зависят. Полученное уравнение состояния такой среды совпадает с уравнением состояния идеального газа "бесструктурных"частиц. Соответственно, одинаковыми будут и термические коэффициенты (растяжения, сжатия, давления). Для определения калорических свойств необходимо точно указать энергетический спектр состояний молекул среды. Чтобы получить качественное представление о величине внутренней энергии и теплоёмкости идеального газа, предварительно укажем значения типичных характеристических температур TX для простейших молекул, где TX = ∆E k и ∆E - разность энергий основного и первого возбуждённого уровней энергии по соответствующим степеням свободы молекулы. Эти значения TX имеют порядок TX ≈ 10K , TX ≈ 103 K , TX ≈ 105 K. Отметим, как это было сделано в п.4.4., что характеристическая температура поступательного движения частицы имеет значение TX ≈ 10−10 K . Для получения последующих выражений используем модели, рассмотренные ранее в п.4.5, а именно, будем считать, что отдельная молекула имеет одну колебательную степень свободы и две вращательные степени свободы. Определим энергию и теплоёмкость системы, связанные с колебаниями молекул. В гармоническом приближении 1 Ei = h̄ω i + , gEi = 1 . 2 Тогда для больцмановской статистической суммы имеем h̄ω Z= e− 2kT h̄ω 1 − e− 2kT . Средняя энергия осциллятора равна E = kT 2 ∂ h̄ω h̄ω ln Z = + h̄ω ∂T 2 e kT − 1 . Для температур T >> Tx = h̄ω k имеем E= h̄ω h̄ω h̄ω + = + kT h̄ω 2 2 1 − (1 − kT + . . .) или E(T ) = kT . 41 Для температур T << Tx получаем E= h̄ω h̄ω 1 + 2e− kT 2 . Выражения для колебательной составляющей энергии системы равны E = NkT для h̄ω T >> Tx и E = N h̄ω 1 + 2 exp − для T << Tx . 2 kT Соответствующие выражения для теплоёмкости системы в этом случае CV = Nk, при N = NA – постоянной Авогадро имеем CV = R (газовая постоянная), 2 h̄ω h̄ω · e− kT CV = Nk · kT . Определим энергию и теплоёмкость системы, связанные с вращательными степенями свободы. Считаем, что для больцмановской статистической суммы имеем (см. ) 2 ∞ h̄ Z = ∑ (2i + 1) exp · i (i + 1) . 2JkT i=0 2 h̄ Для температур T >> Tx = 2Jk получаем Z = TTX . В этом случае получаем для энергии молекулы E(T ) = kT , для энергии системы E = NkT и теплоёмкость CV = Nk или молекулярная теплоёмкость равна R (газовой постоянной). Для температуры T << Tx в статистической сумме определяющую роль играют наTX чальные слагаемые по числу i. Тогда, выбирая i = 0, 1 получаем Z = 1 + 3−2 T . TX TX Представляем ln Z в видеln 1 + 3−2 T ∼ = 3−2 T . Отсюда для энергии молекулы имеем TX E = 6kTX e−2 T . Вращательная составляющая энергии системы в этом случае равна E = N · E и соответствующая теплоёмкость h̄2 CV = 12Nk · 2JkT 2 h̄2 e− JkT . Таким образом, если учитывать поступательное движение молекул, колебательные и вращательные степени свободы для них, что результирующая теплоёмкость системы будет равна CV = Cv +Cv +Cv . Если считать, что между характеристическими температурами существует соотношение TX + TX + TX , то при T > TX результирующая молярная теплоёмкость рассмотренного объекта будет равна 3 7 CV = R + R + R = R. 2 2 Такой результат соответствует только классическому рассмотрению ситуации. Отметим, что в области температур, когда необходимо учитывать квантование энергии (T < TX ), теплоёмкость по соответствующим степеням свободы имеет в основном экспоненциальную зависимость от температуры. 42 При определении полной теплоемкости необходимо также в принципе учитывать составляющую, связанную с электронными возбуждениями молекул. Однако, если считать, что Tx ≈ 104 − 105 K (что типично для простых газовых сред), то величина такой теплоемкости практически равна нулю. Действительно, в этом случае для статической суммы можно записать E0 E0 E1 Z = g0 e− kT + g1 e− kT + ... = g0 e− kT [1 + E1 −E0 E0 g1 g0 e− kT + ...] ≈ g0 e− kT , g0 0 >> T . если считать, что Tx = E1 −E k Тогда получаем для энергии системы E = NE0 , где E0 – энергия основного состояния молекулы, и теплоемкость по этим степеням свободы будет отсутствовать. 4.9 Отрицательные абсолютные температуры Абсолютная термодинамическая температура T является фундаментальной величиной статистической термодинамики. Она определяется феноменологической термодинамике как величина, характеризующая состояние равновесия системы. Математически представляется как производная от энергии системы по ее энтропии, т.е. T = ∂E ∂S . Отсюда следует, что может принимать только положительные значения, если идет речь об обычных термодинамических системах. Следует отметить, что в математическом аппарате феноменологической термодинамики формально можно рассматривать как отрицательную величину. Это не приводит к нарушению функциональных связей между различными величинами, но изменяет их физическое содержание, которое в этом случае не соответствует обычным реальным ситуациям. Статистическая термодинамика вносит дополнительно содержание в представление . Это связано в первую очередь с тем, что по рассчитываемому калорическому уравнению состояния системы E = E(a, T ) можно интерпретировать через энергию системы . В математическом аппарате статистической термодинамики должна быть только положительной величиной, если считать, что реальные физические системы обладают некоторым наименьшим значением энергии 0 , но не имеет верхнего предела энергии, т.е. 0 ≤ n < ∞. В противном случае статистическая сумма (интеграл) Z не будет иметь конечного значения. Единственная возможность рассматривать как отрицательную величину существует для систем, если их энергетический спектр по каким-либо соображениям ограничен сверху, т.е. 0 ≤ n < max . При такой ситуации Z будет содержать конечное число слагаемых и само Z останется конечной, даже если отрицательно. Причина обсуждения таких ситуаций связана с тем, что в настоящее время известны объекты, состояние которых следует представлять отрицательной температурой. Это относится к квантовым усилителям и квантовым генераторам. Отрицательные температуры в таких устройствах возникают, например, по таким представлениям. Допустим, рассматривается система частиц, для которых может существовать только два энергетических уровня. 1 и 2 (2 > 1 ≥ 0). Тогда больцмановская температура такой модели определяется выражением (см. п.4.7.) T= E2 − E1 , 1 k ln N N2 где N1 и N2 в состояниях 1 и 2 соответственно, считаем g1 = g2 , и N1 + N2 = N = Const. Если N1 > N2 , что характерно для нормальных систем, то имеем T > 0. Однако, если 43 удастся реализовать в системе ситуацию, когда N2 > N1 , то мы будем иметь случай отрицательной температуры системы, то есть < 0. Примером такой системы являются N слабо взаимодействующих со средой спино~ Если начальное состояние каждой вых частиц (S = ±1/2) во внешнем магнитном поле H. частицы (полагаем = 0), то в зависимости от направления магнитного момента частицы ~µ вдоль или против поля ее энергия будет равна – µH либо +µH. Рассмотрим некоторые термодинамические свойства такой системы. Статистическая сумма системы равна Z = ZN , Ei µH µH где Z = ∑ gi e− kT = e kT + e− kT = 2ch x, где x = i µH kT . Тогда имеем F = −NkT ln(2ch x), S = N[ln(2ch x) − x], E = −NµH th x. Из анализа этих выражений следует T → +∞, E → −0, S → Nln2, T → −∞, E → +0, S → Nln2, T → +0, E → −NµH, S → 0, T → −0, E → +NµH, S → 0. Таким образом получаем, что система при отрицательных температурах обладает большей энергией. Переход системы из состояния T > 0 в состояние T < 0 происходит при T → ± ∞, т.к. термодинамические величины в этом случае имеют одинаковые значения. 4.10 Дополнительные вопросы 1. Используя методику п. 4.2. получите выражение для термодинамических свойств классического одномерного и двухмерного газа "бесструктурных"частиц. 2. Кинетическую энергию системы для i-ой степени свободы можно представить в виде p2i 1 ∂H = pi , 2m 2 ∂pi где H – функция Гамильтона системы. ∂H Докажите, что 12 pi ∂p = 12 kT, что соответствует теореме о равнораспределении кинетической энергии i теплового движения по степеням свободы классической системы. ∂H Примечание. Результат получается прямым усреднением 12 pi ∂p по функции канонического i распределения. 3. Ориентационная электрическая поляризация дипольных газов. Рассмотрим систему из N свободных частиц, каждая из которых обладает дипольным электрическим моментом ~p. Эту систему помещают во внешнее электрическое поле ~E. Покажите, что величина поляризации ~P среды будет представляться выражением pE kT P = N p cth − . kT pE Примечание. Считать, что состояние каждого диполя во внешнем поле задается функцией классического распределения Больцмана, где E = −~p · ~E. 44 4. Тепловая ионизация атомов. Рассмотрим газовую среду, в которой имеет место однократная ионизация атомов (A0 = A+ + e− ). Такая частично ионизированная плазма находится в объеме V при некоторой температуре T . Считаем, что потенциал ионизации атома равен I. a) Насколько велика ионизация при T ≈ 300 K для простейших газовых сред. b) Покажите, что для любой T выполняется соотношение NA + · Ne − = V · K(T ), NA 0 где mA + · me − kT K(T ) = 2 · 2 mA 0 h 3 2 I e− kT – постоянная равновесия. c) Качественно обсудить температурную зависимость теплоемкости Cv (T ). Точно указать Cv для области низких температур (неионизированный газ) и высоких температур (полностью ионизированный газ). n d) Определить степень ионизации среды коэффициентом α(T ) = nA+0 + nA+ , где nA0 = NA0 N , nA+ A N = NA+ , N = NA0 + NA+ + Ne− . Получить для α(T ) выражение α(T ) = √ 1 1+K(T ) – формула Саха. Примечание. Решение для П.2 основано на использовании для химических потенциалов соотношения µA 0 = µA + + µe − . Молярная теплоемкость двухатомного газа. Рассмотрим двухатомный газ (например N2 ) при температуре T близкой к комнатной. Эта температура достаточно низка, чтобы молекула всегда находилась в низшем колебательном состоянии, но достаточно высока для возбуждения большого числа вращательных состояний. Запишите выражение для среднего значения энергии молекулы в этих условиях. Определите соответствующую молярную теплоемкость Cv газа. Качественно изобразите графическую температурную зависимость Cv (T ) среды для всей области температур (0 < T < +∞). 5 Идеальные системы в равновесном состоянии. Распределение Ферми. Распределение Бозе 5.1 Обобщенное распределение (Гиббса) для системы однотипных частиц Рассмотрим обобщенное распределение (см. п.2.4 и п.3.3) Ω + µN − En,N ωn,N = exp , ∑ ωn,N = 1, kT n,N N – случайная величина, состояние представлено n с соответствующей En,N . Если взаимодействие между частицами отсутствует, т.е система идеальная, то состояние n в общем случае будет определяться набором квантовых чисел ni для каждой частицы n = (n1 , n2 , . . . ni . . . nN ) , 45 а энергия N En,N = En1 ,n2 ,...ni ...nN ,N = ∑ Eni . i=1 Последняя обозначает, что энергия En,N равна сумме энергий Eni отдельных частиц, находящихся в соответствующих состояниях ni . Каждое состояние ni тоже представляется квантовыми числами mi ni = mi1 , mi2 , . . . mij . . . mis , где s – число степеней свободы частицы. В этом случае обозначение вероятности будет записываться в виде ωn,N = ωm1 ,...m1 ,...m1 ,...mN . 1 N s1 sN Предположим, что система состоит из однотипных частиц. Тогда каждое состояние ni будет определяться однотипным набором m j , где 1 ≤ j ≤ s. При такой ситуации есть возможность более просто представлять состояние системы. Действительно, вероятность ωn,N представляет состояние системы в N · s-мерном пространстве квантовых чисел. Теперь, когда рассматриваем однотипные частицы, состояние системы можно задавать в s-мерном пространстве квантовых чисел отдельной частицы. Понятно, что при таком "сжатии"пространства, представляющего состояния системы, в каждом m j окажется число частиц Nm j , которые ранее находились в n1 , n2 , . . . ni . . . nN состояниях. Обобщенное распределение в данном случае приобретает вид Ω + µ ∑sj=1 Nm j − ∑sj=1 Em j Nm j , ωm1 ,...,ms ,N = exp kT где учтено, что химический потенциал µ для всех состояний m j одинаков вследствие равновесного состояния системы, и использованы представления s s j=1 j=1 ∑ Nm j = 1, ∑ Em j Nm j , где Nm j – случайная величина в пространстве квантовых числе s. перепишем выражение для ωn,N в виде ! Ω + ∑sj=1 µ − Em j Nm j ωm1 ,...,ms ,N = exp . kT Омега-потенциал Ω постоянная величина, представляющая систему в данных макроскопических условиях (a, T, µ – постоянные). Тогда запишем ! s µ − Em j Nm j ωm1 ,...,ms ,N = ∏ exp , kT j=1 где C – постоянная. Отсюда получаем выражение для вероятности того, в состоянии m j находится число частиц Nm j (µ − Em ) Nm ωm,Nm = onst exp , kT где индекс " j"опущен, т.к. это выражение справедливо для любого состояния. Условие нормировки имеет вид (µ − Em ) Nm , ∑ ωm,Nm = Const ∑ exp kT Nm Nm 46 " (µ − Em ) Nm Const = ∑ exp kT Nm #−1 ; Z= 1 . Const Суммирование по m в данном случае отсутствует, т.к. рассматривается состояние с заданным m. Для общей функции распределения ωm1 ,...,ms ,N условие нормировки представляется выражением s ∑ ∑ ωm j ,Nm j = ∏ ∑ ωm j ,Nm j = 1. Nm j 5.2 j=1 Nm Распределение Ферми. Распределение Бозе Получим выражение для среднего числа частиц в заданном m состоянии (µ−Em )Nm ∑Nm Nm exp kT ∂ (µ − Em ) Nm Nm = ∑ Nm ωm,Nm = = kT ln ∑ exp . µ−Em ∂µ Nm kT ∑Nm exp m kT Для Ферми частиц Nm = 0, 1. Тогда получаем ∂ (µ − Em ) Nm = kT ln 1 + exp ∂µ kT или 1 ; ∑ Nm = N Nm = (µ−Em ) m exp +1 kT – распределение Ферми. Для Бозе частиц Nm = 0, 1, . . . N. В этом случае имеем ∂ (µ − Em ) (µ − Em ) Nm = kT ln 1 + exp + exp 2 +... . ∂µ kT kT Заменяя выражение в скобках суммой геометрической прогрессии (µ ≤ 0), получаем 1 Nm = exp (µ−Em ) kT ∑ Nm = N ; −1 m – распределение Бозе. 5.3 Критерий применения статистики Больцмана Запишем распределения Ферми, Бозе в виде 1 Nm = exp При условии exp (µ−Em ) kT (µ−Em ) kT . ±1 >> 1 получаем (µ − Em ) Nm = exp kT – распределение Больцмана (часть 4). Критерий применения статистики Больцмана является: 1. Nm << 1, 2. µ < 0 и по величине µ >> kT . 47 5.4 Термодинамические свойства газа ферми-частиц Рассмотрим систему из N свободных "бесструктурных"ферми-частиц в объеме V без внешних полей при температуре T . Считаем T ≈ 0 K (сильное вырождение). В общем случае имеем ∑ Nm = N, ∑ EmNm = E, m m где m определяется квантовыми числами поступательного движения (nx , ny , nz ) и спиновым числом s. Если магнитное поле отсутствует в объеме V , то кратность вырождения по спиновому числу равна gs = (2s+1). В этом случае остается суммирование по квантовым числам поступательного движения. Учитывая, что характеристическая температура, соответствующая поступательному движению мала (см. п. 4.5), состояние частиц можно представлять классически (~r,~p) и суммирование для N и E заменить интегрированием. Тогда получаем для распределения Ферми " N~p = gs ~p2 2m −µ exp kT #−1 , а для числа частиц N и энергии системы выражения 2 R V d~p g [exp ~p /2m−µ + 1]−1 = N kT h3 s R ~p2 /2m−µ V d~p −11 E(~p) h3 gs [exp kT + 1] = E. Рассмотрим распределение функции Ферми f (E) при ≈ 0К E −µ 1, E ≤ µ0 −1 f (E) = [exp + 1] = kT 0, E µ0 Тогда уравнение для N и E приобретет вид ZPF ZPF V d~p gS = N, h3 E(~p) 0 V d~p gS = E, h3 0 где PF –максимальное значение импульса частицы (импульс Ферми). В этих выражениp2 ях учтем E(~p) = 2m , d~p = 4πp2 d p. Тогда получаем 4πV gS N= h3 ZPF p2 d p, = 4πV gS 3 p 3h3 F 0 Отсюда имеем для импульса Ферми (PF ) pF = ( 3N 1/3 ) h, 4πV gS для энергии Ферми (EF = µ0 ) µ0 = EF = p2F 3N 2/3 h2 =( ) 2m 4πV gS 2m 48 Полная энергия системы частиц равна V gS E= mh3 ZpF p4 d p = 2πV gs 5 p 5mh3 F 0 – калорическое уравнение состояния, ( ≈ 0К) Представленные выражения можно достаточно надежно использовать не только при ≈ 0 К, но и при любой температуре , при условии µ >> k. Рассмотрим случай > 0 (слабое вырождение). Задача в этом случае сводится к вычислению интегралов вида Z∞ E n dE In = E−µ e kT + 1 0 При условии kT µ << 1 выражение для I приближенно имеет вид 4 In = µn+1 π2 n(n + 1) kT 2 [1 + ( ) ] n+1 6 µ Тогда выражение для N имеет вид 2π(2m)3/2 gSV N= h3 где Z∞ E 1/2 dE 0 e E−µ kT , +1 2 π2 kT I1/2 = µ3/2 [1 + ( )2 3 8 µ Подстановка приводит к выражению N= 2π(2m)3/2 gSV 2 3/2 π2 kT 2 × µ [1 + ( ) ] h3 3 8 µ При ≈ 0 К отсюда имеем для µ0 µ0 = ( 3N 2/3 h2 ) , 4πV gS 2m что соответствует ранее полученному. Приравнивая NT >0 и NT ≈0 получаем π2 kT 3/2 µ0 = µ 1+ ( ) . 8 µ Выражая µ, получаем зависимость π2 kT 2 µ(T ) = µ0 1 − ( ) , 12 µ0 где использовано представление биномиального ряда (1 − x)M = 1 − mx + ..., и в разложении осуществлена замена µ на µ0 . 4 См Ю.Б. Румер, М.Ш. Рывкин, Термодинамика, статистическая физика и кинематика, Наука., М.,1977. 49 Аналогичные расчеты можно провести для энергии системы. Результат можно привести к виду 5π2 kT 2 3 E(T ) = Nµ0 [1 + ( ) ]. 5 12 µ0 Теплоемкость системы равна π2 kT cV = Nk( ). 2 µ0 Для определения других термодинамических характеристик надо использовать более общий подход. Определим термодинамический потенциал Ω для исследуемой системы. Из п.5.2. следует µ − Em Ω = −kT ∑ ln(1 + exp ) kT m Если перейти к интегрированию по классическим переменным (~r,~p), то получим √ Z∞ 4π 2(m)2/3 gSV µ − E 1/2 Ω = −kT ln(1 + exp )E dE 3 h kT 0 Интегрируя по частям приходим к результату Z∞ µ − E 1/2 2 µ − E 3/2 ∞ 2 ln(1 + exp )E dE = ln(1 + exp )E |0 + kT 3 kT 3kT 0 Z E 3/2 dE e E−µ kT + 1 , где первое слагаемое равно нулю на обоих пределах, а второе интегральное слагаемое определяет энергию системы E. Тогда имеем 2 Ω = − E, 3 где √ Z 3/2 4π 2m3/2 gSV EP dE p E= E p −µ h3 e kT + 1 – калорическое уравнение состояния. Соотношения между калорическим и термическим уравнениями состояния представляются в виде pV = 23 E, где Ω = −pV . Последующий расчет связан с определением интеграла I3/2 . Приближенное выражение для этого случая приведено выше. Тогда термическое уравнение состояния имеет вид 2N 5π2 kT 2 p= µ0 1 + ( ) , 5V 12 µ0 где при = 0 имеем нулевое давление 1 p0 = 5 3 8π 2 5 3 N 3 V Для рассматриваемой модели можно в более конкретном виде, чем ранее (п.3.3), определить условия перехода при исследовании свойств к статистике Больцмана. Таким критерием ранее было µ < 0 и |µ| >> (см п.5.3). 50 Возьмем для µ выражение для энергии Ферми EF = µ0 и положим µ0 = . Отсюда имеем, используя выражение для µ0 TB = 3N 2/3 h2 ( ) 2mk 4πV gS – температура вырождения. Тогда при < для данной модели исследование ее свойств возможно только с использованием распределения Ферми. При >> расчет характеристик можно проводить, используя распределение Больцмана. Следует отметить, что определить температуру вырождения для данной модели можно, используя представления больцмановской статистики. Действительно, химический потенциал µ для данной модели в больцмановском представлении (см. п.4.2) имеет вид µ = kT ln Nh3 (2πmkT )3/2V Требование µ < 0 и |µ| >> kT позволяют записать выражение для определения температуры Nh3 = (2πmkT )3/2V Из этого имеем f rac32 h2 N TB = 2πmk V , что с точностью до постоянных величин, ненамного отличающихся от единицы, это выражение совпадает с ранее полученным для из распределения Ферми. Можно дать квантовомеханическое представление условия применения статистики Больцмана. Известно, что для волны де Бройля можно записать λD = h̄p , где –импульс частицы. Обозначим через r0 среднее расстояние между частицами системы. Тогда при r0 >> λD квантовомеханическое взаимодействие между частицами отсутствует и систему следует рассматривать как классическую. Это выполнимо при условии λrD0 1. Для 1 последующих оценок ситуации воспользуемся выражениями p = (2mkT ) 2 – тепловой 1 1 импульс частицы поступательного движения, r0 = VN 3 = n− 3 , где n – концентрация частиц. Отсюда следует, что классическое приближение справедливо, если выполняется 1 h̄ √ n 3 << 1. 2mkT Отметим, что получаемая из этого из этого выражения температура вырождения соответствует результатам при ее термодинамическом определении. 5.5 Термодинамические свойства газа бозе-частиц Рассмотрим систему из N свободных бозе-частиц в объёме V без внешних полей при температуре T . Для такой модели применяем распределение Бозе в виде (см. п. 5.2) 1 N= e Em−µ kT −1 , ∑ Nm = N, m где состояние m представляется квантовыми числами (n1 ,n2 ,n3 ) поступательного движения и спиновых числом s. Отметим особенность распределения по энергетическому спектру для частиц-бозе (по сравнению с частицами ферми). При некоторой температуре T должно иметь место 51 распределение частиц по всем состояниям Em (0 ≤ Em < ∞). При T → 0 K все N частиц должны находиться в состояниях с минимальной энергией Em (Em = 0). Далее, для такой системы должен существовать интервал температур 0 < T < TB , для которого химический потенциал должен ровняться 0 (µ = 0). Действительно, из распределения Nm (Em = 0) следует, что µ → − kT N и при T → 0 имеем для µ → −0. В то же время, эта модель в больцмановской статистике (см. п. 4.2) в термодинамическом пределе имеет конечное значение для химического потенциала µ(T ). В связи с этим распределение для Nm следует представлять в виде −1 T < TB Nm = exp EkTm − 1 h i−1 Nm = exp EmkT−µ T > TB , где TB – характеристическая температура для системы, имеющая смысл температуры вырождения (см. п. 5.4). Получим выражение для TB . Для этого в условии нормировки для Nm перейдём от суммирования по квантовым состояниям к интегрированию по классическим переменным (~r,~p), имея ввиду, что характеристическая температура (Tx ) поступательного движения чрезвычайно мала. Тогда находим 1 √ 3 Z∞ E p2 dE 4π 2m 2 gsV N(T ) = , E p −µ h3 e kT − 1 0 где gs – кратность вырождения по спину частицы, E p – кинетическая энергия частицы. При T = TB полагаем µ = 0 и получаем уравнение для определения TB , считая NT ∼ =N 1 √ 3 Z∞ E p2 dx 4π 2m 2 gsV N(T ) = . Ep h3 kT 0 e B −1 Отсюда, используя представления интегралов вида Z∞ xn dx , ex − 1 0 находим TB Z∞ 1 x 2 dx = 2, 33; ex − 1 0 Z∞ 3 x 2 dx = 1.78. ex − 1 0 2 2 3 N h √ TB = · , mk 4π 2gsV · 2, 33 что с точностью до численных коэффициентов совпадает с ранее полученными выражениями для температуры вырождения (см. п. 5.4). Запишем выражения для числа частиц N(T ) и полной энергии системы E(T ), зависящих от температуры T , при T < TB . N(T ) = E(T ) = √ 3 ∞ 1 4π 2m 2 gsV R E p2 dE Ep h3 0 e kT −1 √ 3 ∞ 3 4π 2m 2 gsV R E p2 dE Ep h3 0 e kT −1 52 √ 3 3 4π 2m 2 gsV = 2, 33 (kT ) 2 , h3 √ 3 5 4π 2m 2 gsV = 1, 78 (kT ) 2 , h3 где E(T ) – уравнение состояния (при T < TB ). Используя представление функции распределения для частиц –бозе (см. п. 5.2.), получают выражения для омега-потенциала в виде Ω = 32 E, где E – полная энергия системы. Если учесть Ω = −ρV , то имеем pV = 23 E. Отсюда термическое уравнение состояние бозе-газа для T < TB определяется формулой √ 3 5 2 4π 2m 2 gs p = 1.78 (kT ) 2 . 3 3 h Таким образом давление p не зависит от объёма V и стремиться к 0 при низких температурах. Последнее обстоятельство связано с тем, что основная часть частиц при таких условиях находятся в состоянии E0 = 0 и не обладает импульсом, определяющим давление. 5.6 Статистика электронов в металлах и полупроводниках При исследовании свойств валентных электронов в металлах в зависимости от решаемых проблем используют для них различные модельные представления. Термодинамические свойства в достаточной степени могут быть рассмотрены в представлении свободных и независимых электронов (модель Зоммерфельда). Это предполагает, что между электронами отсутствует взаимодействие и электроны способны свободно перемещаться в однородном потенциальном поле кристаллической решетки. Такое представление валентных электронов в металлах соответствует модели идеального бесструктурного газа ферми-частиц (см. п. 5.4) со спином s = 1/2 и кратностью вырождения gs = 2. Оценим величину температуры вырождения TB валентных электронов h2 TB = 2m∗ k 3N 8πV 2 3 , где m∗ – эффективная масса электрона. Используя типичные значения для m∗ и n = VN , получаем для TB = 104 K. Таким образом валентные электроны при всех допустимых температурах следует рассматривать как сильно вырожденную систему ферми частиц. Оценим величину энергии Ферми EF = µ0 . Используя представление µ0 (см. п. 5.4), получаем µ0 = 5 эВ. Отсюда следует, что химический потенциал µ(T ) валентных элек kT ∼ тронов при T = 300 K практически соответствует энергии Ферми, т.к. отношение µ0 = 10−2 при kT = 0.025 эВ. ∼ −2 с Аналогично, полная энергия валентных электронов при отношении kT µ0 = 10 достаточной степенью точности будет соответствовать значению их энергии при T = 0 K. Приведём дополнительно значения величин - давление (T = 0 K) p ∼ 104 − 105 атм., - скорость электронов v ∼ 108 cм/c. Свободные электроны при любой температуре (T > 0) способны покинуть объём металла. Наиболее существенно появление при больших температурах (термоэлектронная эмиссия). Выражение для концентрации термоэлектронов имеет вид √ 3 Z∞ 1 8π 2m 2 V E 2 dE , NT = E+E0 −µ h3 e kT + 1 0 53 где, положим gs = 2, E0 – величина потенциального барьера для валентного электрона в металле, т.е. при E = E0 валентный электрон покидает объём металла с нулевой кинети0 −µ ческой энергией. Если считать EkT >> 1, то получаем √ 3 √ 3 3 Z∞ 3 (E0 −µ) E 1 8π 2m 2 V − E0 −µ 4 2π 2 m 2 − 2 e kT kT kT E 2 dE = NT = e e (kT ) h3 h3 0 R∞ √ π 2 1 где учтено, что интеграл ex x 2 dx = 0 (см. Приложение П.4.2). Величину (E0 − µ) = AB – называют работой выхода электрона из металла. Она имеет порядок в несколько электрон-вольт. Моделирование валентных электронов в полупроводниках, также как и описание их свойств является более сложной задачей, чем у металлов. Продемонстрируем это на примере собственного полупроводника. Рассмотрим валентные электроны, которые могут находиться в валентной зоне (V-зона) и зоне проводимости (C-зона) полупроводника. Условие нормировки для них представляется в виде N=∑ i gi e Ei −µ kT +1 +∑ j gj e E j −µ kT +1 = ∑ gi , i где индексом i обозначены уровни энергии V-зоны, индексом j – уровни энергии в Cзоне, gi и g j – кратности вырождения. При достаточно высоких температурах, например, T ≈ 300 K число электронов в Cзоне для наиболее типичных полупроводников (Ge, Si, GaAs) относительно невелико (Nec << N) и их следует рассматривать как невырожденную подсистему. В то же время, электроны Nev в V-зоне практически полностью заполняют энергетические уровни и статистически их следует рассматривать как вырожденную подсистему. Условие нормировки для всей системы электронов в этом случае можно представить в форме N=∑ i gi e Ei −µ kT +1 + ∑ g je µ−E j kT . j Такое выражение, использующее одновременно два распределения Ферми и Больцмана в данном случае и в последующих расчетах является неудобным. Для исключения этой ситуации вводится понятие второго типа носителей заряда в полупроводнике – дырки, как незаполненное состояние электрона в V-зоне. В этом случае для V-зоны носителями заряда будут являться дырки, а для C-зоны носители – электроны. При этом эти два типа частиц будут представляться всегда одним и тем же распределением, характерным для невырожденных или вырожденных систем. Установим функцию распределения для дырок из условия ! gj gi 1 . = ∑ µ−Ei ∑ E j −µ = ∑ gi 1 − Ei−µ i e kT + 1 j e kT + 1 i e kT + 1 Таким образом распределение Ферми для дырок имеет вид 1 Ni = e µ−Ei kT , +1 54 0 ≤ Ni ≤ 1 , которое при exp µ−Ei kT >> 1 переходит в распределение Больцмана Ni = e Ei −µ kT . Получим выражение для числа электронов Nec в C-зоне и числа дырок Nhv в V-зоне в собственном невырожденном полупроводнике. Энергетический спектр состояний в зонах считаем непрерывным. Тогда записываем ZEC 2V NeC = 3 h 0 2V NhV = 3 h 1 e E−µ kT −∆E Z EV −p , d→ +1 1 e µ−E kT −p , d→ +1 где начало отсчета энергии поместили на дне зоны проводимости. Для дальнейшего расчёта необходимо знать статистический вес g(E) электронов и дырок. Будем считать, что p2 для них выполняется соотношение между импульсом и энергией в виде E = 2m . В этом случае получаем √ 3 1 Z∞ E 2 dE 8π 2 (m∗e ) 2 V C , Ne = E−µ h3 e kT + 1 0 √ 3 Z 1 ∗ 2 V −∆E 8π 2 m (−∆E − E) 2 dE h V Nh = , µ−E h3 kT + 1 e −∞ где пределы в интегралах Ec и Ev – заменены на +∞ и −∞, а через m∗e и m∗h обозначены эффективные массы электрона и дырки. µ−E v Будем считать, что для Nec выполняется E−µ kT >> 1, а для Nh выполняется kT >> 1. Тогда подынтегральные выражения можно заменить экспонентами. Для дальнейшего E расчёта используем замену x = kT и x = −∆E−E kT . Тогда получаем 3 √ Z∞ 1 2 3 ∗ µ 8π 2 (me ) V 2 C (kT ) e kT x 2 e−x dx, Ne = 3 h 0 √ 32 Z∞ ∗ 3 1 8π 2 mh V 2 − µ+∆E V −x 2 kT Nh = (kT ) e e x dx, h3 0 R∞ где e−x x f rac12 dx = 0 √ π 2 (см. Приложение П.4.1). Окончательно для Nce и Nvh записываем NeC NhV 2πm∗e kT =2 h2 2πm∗h kT =2 h2 55 3 2 µ e kT , 3 2 e− µ+∆E kT , Из равенства Nec и Nhv получаем ∗ mh ∆E 3kT . µ=− + ln 2 4 m∗e По рассмотренной схеме расчёта не представляет труда установить полную энергию валентных электронов и их теплоёмкость. (см. Дополнительные вопросы. П.7.). 5.7 Равновесное тепловое излучение Любой вещественный объект является источником теплового электромагнитного излучения. Термин "тепловое"означает, что электромагнитное поле возникает за счет внутренней энергии объекта. Рассмотрим частный случай теплового излучения, когда оно возникает, например, в полости вещества, температура которого . Тогда и полость, в которой существует излучение, следует характеризовать такой же температурой. Равенство температур, соответствующее равновесному состоянию, свидетельствует, что перераспределения энергии между веществом и полем в термодинамическом смысле не происходит. Будем рассматривать тепловое равновесное излучение как фотонный газ. Известно, что масса покоя фотона равна нулю (m0 = 0). Энергия и импульс фотона связаны соотношением E = pc, где – электродинамическая постоянная. Фотоны имеют целочисленный спин, равный единице. Соответственно они относятся к классу базе-частиц. Кроме того, фотонный газ следует рассматривать как идеальную систему, т. к. электромагнитное поле представляется линейными дифференциальными уравнениями (Максвелла). Для описания термодинамических свойств такой системы будем использовать результаты, полученные для абстрактной модели газа базе-частиц(см. п. 5.5.). Температура вырождения (TB ) зависит от массы частицы m0 . Для m0 = 0 получаем бесконечно большое значение TB . Отсюда следует, что тепловое равновесное излучение следует рассматривать как сильно вырожденную базе систему, для которой химический потенциал µ = 0. Распределение Базе в данном случае имеет вид 1 N̄~k = e E~ k kT −1 , где состояние представлено волновым вектором~k = 2π λ ~n0 , где~n0 – волновой вектор. Импульс ~p и волновой вектор связаны соотношением ~p = h̄~k. Для макроскопических объемов V теплового излучения величину ~k следует рассматривать как непрерывную. Запишем распределение Базе в энергетическом представлении, используя выражение E = h̄ω 2 , N (ω) = h̄ω e kT − 1 где множитель "2"учитывает два направления поляризации колебаний поперечной электромагнитной волны (gω = 2). Для равновесного числа фотонов в интервале частот ω − ω + dω воспользуемся формально представлением, полученным ранее (см. п. 5.5.) d~r · d~p . 3 −1 h gs dN (~r,~p) = e E kT 56 или dN (~p) = V gs d~p , h3 e kTE − 1 где d~p = 4πp2 d p. В данном случае после перехода от импульса ~p к частоте ω получаем dN (ω) = V ω2 dω . π2 c3 e h̄ω kT − 1 Полное число равновесных фотонов N в объёме V при температуре T определится выражением 3 Z∞ 2 V ω dω V kT N= 2 3 = 2, 40 2 3 , h̄ω π c π c h̄ e kT − 1 0 где использовано представление Z∞ x2 dx = 2, 40. ex − 1 0 Спектральное распределение энергии в тепловом равновесном излучении имеет вид dE (ω) = h̄ω · dN (ω) = Vh̄ ω3 dω . π2 c3 e h̄ω kT − 1 Максимум кривой распределения энергии соответствует частоте ωmax = Const · T , где Const ∼ = 2, 82. Рассмотрим спектральное распределение при низких (инфракрасных) и высоких(ультрафиолетовых) частотах. Считаем h̄ω << kT . Тогда, используя разложение eX = 1 + X + ..., получаем dE (ω) = kT · V 2 ω dω π2 c3 – формула Рэлея-Джинса. Считаем h̄ω >> kT . Тогда, пренебрегая в знаменателе единицей, получаем dE (ω) = Vh̄ 3 − h̄ω ω e kT dω π2 c3 – закон Вина. Полная энергия излучения равна Vh̄ E= 2 3 π c Z∞ h̄ω 0 π2V k4 4 = T 3 − 1 15c3h̄ ω3 dω e kT – закон Стефана-Больцмана, где при вычислении использовано представление Z∞ x3 dx π4 = . ex − 1 15 0 Выражение для следует рассматривать как калорическое уравнение состояния системы. 57 Можно показать, что Омега-потенциал теплового равновесного излучения представляется через энергию системы в виде 1 Ω=− E 3 (см. Дополнительные вопросы. П. 3). Тогда имеет 1 pV = E 3 . Соответственно термическое уравнение состояния равновесного излучения имеет вид 1 π2 k 4 4 · T . 3 15c3h̄3 p= 5.8 Метод наиболее вероятного распределения Обсудим универсальный метод получения равновесных функций распределения. Этот метод основан на использовании выражения S = k lnW , где S – энтропия системы, W – набор микросостояний системы (статистический ансамбль). Содержание этого выражения обсуждалось ранее (см. п. 2.1.). В равновесном состоянии S имеет максимальное значение (см. п. 1.). Тогда из анализа S на экстремальное значение можно установить функцию распределения для микросостояний системы, находящейся в равновесном состоянии. Покажем, что с помощью такой методики достаточно просто получить распределения Больцмана, Ферми, Базе. Основная задача сводится к получению аналитического выражения для W . Для идеальных систем из однотипных частиц можно поступить следующим образом. Каждое состояние Ei частицы можно представить "энергетической полкой", число ячеек которой равно соответствующей кратности вырождения gi . Тогда энергетический спектр частицы представится набором таких "полок". Если в системе N частиц, то, размещая их по "полкам"и "ячейкам", можно установить "W". Отметим особенности размещения для частиц трех тиров (больцмановских, ферми, бозе). Рассмотрим простейший случай. Разместим две частицы по трем ячейкам (Ni = 2, gi = 3). Для различных частиц a и b имеем девять различных размещений. Для частиц-базе вследствие их неразличимости возможно шесть размещений. Для ферми-частиц вследствие их неразличимости и принципа Паули возможно только три размещения. Используя методы комбинаторики, получаем выражения для W , размещая N частиц по "полкам"i и "ячейкам"gi . Имеем: различимые частицы i gN W = N!∏ i , i Ni ! частицы бозе W =∏ (Ni + gi − 1)! , Ni !(gi − 1)! W =∏ gi ! , Ni !(gi − Ni )! i частицы ферми i 58 где Ni - число частиц в состоянии Ei . Эти выражения определяют полное число состояний. Для термодинамических систем энтропия S определяется числом доступных состояний. Условие нахождения системы в доступных состояниях можно сформулировать следующим образам. Будем считать, что заданы полное число частиц и полная энергия системы, т.е. ∑ Ni = N, ∑ Ni Ei = E. i i В этом случае искомой результат получают, используя анализ на условный экстремум (метод Лагранжа). Получим распределение, соответствующее различимым частицам. Имеем в данном случае( постоянную к – Больцмана опускаем) i gN S = lnW = ln N! + ∑ ln i = ln N! + ∑ (Ni ln gi − ln Ni !) =N ln N + ∑ (Ni ln gi − Ni ln Ni ), Ni ! i i i где использовано (формула Стирлинга) ln N! = N ln N − N ∼ = N ln N. Определяем функцию Φ, учитывающую условия (N, E – заданы) Φ = S−αN−βE, где α и β – постоянные множители. В развернутом виде имеем Φ = N ln N + ∑ (Ni ln gi − αNi − βNi Ei ). Рассматриваем экстремальное значение функции Φ dΦ = [ln gi − ln Ni + (1 − α) − βEi ] = 0. dNi ∑ Отсюда получаем ln Ni − ln gi = (1 − α) − βEi 59 или Ni = Ce−βEi , gi где постоянная C = exp(1 − α). Постоянные C и β устанавливаются из условий, представляющих состояние системы (в данном случае заданы N и E). Если в качестве конкретного объекта использовать идеальный газ частиц, то будем иметь Z N = N(~r,~p)dxdydz4πp2 d p, Z E= p2 N(~r,~p)dxdydz4πp2 d p, 2m где p2 N(~r,~p) = Ce−β 2m . Эти два уравнения позволяют устанавливать и β, если E = 23 NkT . Отсюда получаем 1 β = kT и N C= . (2πmkT )3/2V 5.9 Дополнительные вопросы 1. Считаем, что значению длины волны де-Бройля частицы отвечает выражение λd = √ h̄ , 2mkT где m – масса частицы, T – температура, а среднее расстояние между частицами r0 ∼ 1 n− 3 , где n – концентрация частиц. Сделайте численные оценки λd и r0 для двух сред. а) Газообразный гелий при комнатной температуре (T = 300 K) и атмосферном давлении = 760 мм. рт. ст. б) Валентные электроны в типичном металле, например меди. Внешние условия те же, что и в п. 1.1. Используя критерий применимости классической статистики (Больцмана) в виде r0 >> λd , выясните условия его выполнения для этих двух сред. 2. Рассматривается газ бозе-частиц. Омега-потенциал системы представляется в виде Ω = ∑ Ωm , где Ωm – потенциал частицы N. Известно (см. п. 5.2.), что Ωm = m m kT ln(1 − exp µ−E kT ). Применяя методику п. 5.4., покажите, что выполняется соотношение Ω = −(2/3), где – полная энергия системы. Убедитесь, что это выражение сохраняется и для случая µ = 0. 3. Рассматривается тепловое равновесное излучение. Пока жите, что выражение связывающее полную энергию и омега-потенциал Ω системы, представляется в виде Ω = −(1/3)E. Примечание. Использовать решения п. 2. 4. Распределение Ферми и Бозе для идеализированной, модели. Применяя обобщенное распределение (Гиббса) , установить выражение для чисел заполнения, энтропию, теплоёмкость и химический потенциал газа с двумя энергетическими уровнями для частиц 1 и E2 , где E2 > E1 , и одинаковыми кратностями вырождения g1 = g2 в случае ферми- и бозе-частиц. 60 5. Качественно графически изобразите температурную зависимость химического потенциала µ(T ) и полной энергии E(T ) газа ферми-частиц и бозе-частиц для интервала температур включающего температуру вырождения (T) . 6. Оцените величину температуры вырождения (T) для типичной газовой среды (n ≈ 19 10 см−3 ) и электронного газа при n ≈ 1022 см−3 (металлы) и при n ≈ 1010 см−3 (полупроводник, например Si пи ≈ 300 К). 7. Используя результаты п. 5.6., получите выражения для теплоемкости V собственного невырожденного проводника. 8. Запишите выражения для концентрации электронов и дырок в примесном полупроводнике с концентрацией донорной примеси Nd = 1010 см−3 ; 1020 см−3 при условии ∆g >> ∆Ed , ∆Eg >> , >> ∆d , где ∆g – запрещенная зона полупроводника, ∆d – энергия активных примесей. 9. Тепловое равновесное излучение находится в объеме V , в котором одновременно присутствует газовая среда. Получится выражение для температуры , при которой объемная плотность энергии газовой среды будет равна объемной плотности энергии излучения. Оценить величину , если плотности частиц газовой среды соответствует нормальным условиям n ∼ 1019 cм−3 . 10. Максимизация энтропии (дополнение к п. 5.8.). Система может находится в любом из N состояний. Вероятность того, что система находится в i-ом состоянии равна ωi (i = 1, 2, . . . , N), причем ∑ ωi = 1. Применяя метод неопределенных множителей (Лагранi жа) показать, что распределение вероятности, соответствующее максимуму энтропии, представляется общим выражением S = −k∑ ln ωi ωi , i имеем вид ω1 = ω2 = . . . = ωN = 1/N и для энтропии в данном случае выполняется S = Smax = k ln N. 6 Равновесные ТД системы взаимодействующих частиц Рассматриваются системы, для которых имеет место взаимодействие между частицами Если система состоит из N однотипных частиц, то функция Гамильтона для неё в общем виде представляется выражением N H (X) = ∑ i=1 ~p2i + E0 (~ri ) + E (~r1 ,~r2 , ...~rN ) , 2m (87) Где первое слагаемое определяет кинетическую энергию теплового движения частиц и их потенциальную энергию во внешнем поле, второе слагаемое соответствует потенциальной энергии взаимодействия частиц. равновесных Для равновесных систем их состояние представляется с помощью функций распределения на всю систему (Гиббса), например, функцией канонического распределения (см. п. 2) 61 1 H(X) 1 En f (X) = e− kT ωn = e− kT . (88) z z Последующий статистический расчет возможен, если точно указана функция H (X) и установлен спектр собственных значений En системы. 6.1 Конфигурационный интеграл. Частичные функции распределения Будем считать, что система состоит из N свободных "бесструктурных"частиц в объёме V без внешних полей, E0 (~r) = 0. Определим статистический интеграл Z классического канонического распределения. В общем случае имеем Z Z= e− H(X) kT dX . N!h3N (89) Если использовать выше представленное выражение для H(X), то получим 1 z = z N QN , V Z QN = e− где E (~r1 ,~r2 ,...~rN ) kT d~r1 ...d~rN , (90) 3N (2πmkT ) 2 ·V N z= . (91) N!h3N Выражение для QN представляет конфигурационный интеграл системы. При таком представлении Z задача сводиться к установлению QN . Действительно, для свободной энергии F имеем F = −kT ln z. В данном случае получаем (92) F = F+ F, где F – свободная энергия системы в предположении, что она идеальная, F = QN −kT ln V N – часть свободной энергии, определяемая взаимодействием. Для дальнейших расчетов требуется моделирование характера взаимодействия между отдельными группами из s-частиц, то потенциальная энергия для такой группы принимает более простой вид E (~r1 , ...~rs ). Для парного взаимодействия между частицами функция Гамильтона будет иметь вид N N ~p2i + ∑ Ei j ~ri ,~r j , H (X) = ∑ i, j=1 i=1 2m (i 6= j) , (93) где Ei j ~ri ,~r j -потенциальная энергия взаимодействия i-ой и j-ой частицы. Более общий подход в таких случаях основан на использовании представления состояния системы с помощью частичных функций распределения fs (x1 , x2 , ...xs ), где xi = (~ri ,~pi ). Эта функция распределения определяет состояние группы из s частиц. В то же время она должна быть представлена таким образом, чтобы с её помощью были установлены свойства всей системы в целом. Установление конкретного вида частичных функций распределения проводиться двумя способами. Решается отдельная задача по установлению функций распределения для исследуемой модели либо она выражается через 62 известную, в качестве которой для равновесных термодинамических систем можно использовать одну из функций распределения Гиббса. Рассмотрим соотношения, которые должны выполняться для функций распределения. Все частичные функции распределения представляются через функцию канонического распределения (Гиббса). fs (x1 , ...xs ) = V s Z f (X) dxs+1 ...dxN , 1 fs dx1 ...dxs = 1. Vs Между частичными функциями распределения выполняется соотношение Z 1 fs+1 dxs+1 . fs = V В свою очередь можно представить выражение Z (94) (95) (96) N f (X) = f (~r1 ,~r2 ...~rN ) ∏ f (~pi ), (97) i=1 где f (~pi ) – одночастичная функция распределения Максвелла, 1 − E (~r1 ,~r2 ...~rN ) kT e QN – функция распределения по координатам. Для неё опять можно записать f (~r1 ,~r2 , ...~rN ) = f (~r1 ,~r2 , ...~rs ) = V и s Z f (~r1 ,~r2 , ...~rN ) d~rs+1 ...d~rN (98) (99) 1 fs (~r1 ,~r2 , ...~rs ) d~r1 ...d~rs = 1. (100) Vs Установим конкретную структуру, например, для двухчастичной функции распределения f2 (x1 , x2 ) в классическом представлении. Как следует из приведенных выражений Z E12 f2 (x1 , x2 ) = Ce− kT f1 (~p1 ) · f2 (~p2 ) , (101) где E12 (~r1 ,~r2 ) – потенциальная энергия парного взаимодействия, f1 (~p1 ) и f2 (~p2 ) – распределение Максвелла по импульсам. Постоянная находится из условия нормировки Z 1 f2 (x1 , x2 ) dx1 dx2 = 1. (102) V2 Тогда имеем Z −1 E12 1 − kT C= e d~r1 d~r2 . (103) V2 Для какой модели термодинамические величины системы предоставляются через одночастичную f1 (x1 ) и f2 (x1 , x2 ) двухчастичную функцию распределения. Действительно, для полной кинетической энергии системы имеем N Ek = H X~p · f (X) dX = V Для полной потенциальной энергии получаем Z Z E= H (X~r ) · f (X) dX = Z N ∑ Ei j · f (X) dX = i, j=1 63 Z ~p2 f1 (x) dx. 2m N (N − 1) 2V 2 Z (104) E12 · f2 (x1 , x2 ) dx1 dx2 . (105) 6.2 Термическое уравнение состояния газовой среды с парным взаимодействием Рассмотрим среду из N частиц вобъёме V , между которыми имеет место парное взаимодействие. Обозначим E ~ri ,~r j - потенциальную энергию парного взаимодействия. Потенциальная энергия системы равна E = ∑ E ~ri ,~r j . i< j Получим выражение для свободной энергии системы, связанной со взаимодействием частиц (см. п. 6.1) Z E 1 F = −kT ln N e− kT d~r1 ...d~rN . (106) V Представим подынтегральное выражение в виде h E i E (107) e− kT = 1 + e− kT − 1 . Тогда имеем Z E 1 − kT F = −kT ln 1 + N e − 1 d~r1 ...d~rN . (108) V Предположим, что подынтегральное выражение можно заменить суммой по всем парам (доказательство такой замены приведено А.И. Ансельм., Основы стат. физики и ТД, М.,Наука, 1973. Ю.Б. Румер, М.Ш. Рывкин, Термодинамика, стат. физика и кинетика, М.,Наука,1977.). " # E (~ri ,~r j ) E e− kT − 1 = ∑ e− kT − 1 . (109) i< j Получаем Z E (~r ,~r ) 1 2 N (N − 1) − kT F = −kT ln 1 + e − 1 d~r1 d~r2 . (110) 2V 2 Будем считать, что взаимодействие определяется только r-взаимным расположением частиц (r = |~r1 −~r2 |). Выражение для F приводится виду Z N 2 − E(~r) F = −kT ln 1 + e kT − 1 d~r . (111) 2V Обозначим A (T ) = 1 2 Z 1 − e− E(~r) kT d~r. (112) Получаем выражение N2 F = −kT ln 1 − A (T ) . (113) V Будем считать, что имеем слабое отклонение (|E (r)| << kT ) от идеального состояния. Осуществляя разложение логарифма в ряд, в этом случае приходим к выражению N2 A (T ) . V Термическое уравнение состояния рассмотренной модели имеет вид ∂F N2 N N p=− = kT + kT 2 A (T ) = kT 1 + A (T ) . ∂V T V V V F = kT (114) (115) Где первое слагаемое соответствует уравнению состояния идеального газа. Дальнейшее уточнение уравнения p(T,V ) связанно с определением коэффициента A(T ), что рассматривается в дополнительных вопросах (п. 6). 64 6.3 Термодинамические свойства кристаллических тел Рассматриваем свойства, связанные с тепловым движением частиц, образующих кристаллические тела (диэлектрики, металлы, полупроводники). Для таких объектов можно выделить две подсистемы, формирующие их термодинамические свойства - кристаллическую решетку и электронный газ. Термодинамические свойства валентных электронов в металлах и полупроводниках обсуждались ранее (см. п. 5.6). Получим выражение для теплоёмкости кристаллической решётки. Исторически существуют три подхода к решению этого вопроса. Кристаллическая решетка моделировалась системой классических осцилляторов (классическая теория), системой квантовых однотипных осцилляторов (теплоёмкость по Эйнштейну) и системой связанных квантовых осцилляторов (теплоёмкость по Дебаю). Теория теплоемкости по Эйнштейну приведена в дополнительных вопросах (п.6). Рассмотрим теплоёмкость кристаллической решётки в представлении теории Дебая. Будем считать, что частицы (атомы, ионы, молекулы), образующие кристаллическую решетку, совершают гармонические колебания. Так как между частицами существует взаимодействие, то такие колебания осцилляторов следует рассматривать как связанные. В таком случае в системе связанных осцилляторов должны устанавливаться совместные (нормальные) колебания с разными частотами. Полное число нормальных колебаний для кристалла из N-частиц равно 3N. Если учесть, что N ∼ = 1022 cm−3 , то следует считать спектр нормальных колебаний непрерывным. При этом каждое нормальное колебание происходит так, как если бы остальных не было. Таким образом кристаллическую решетку следует рассматривать как идеальную систему осцилляторов с непрерывным спектром нормальных частот для них. Полную энергию тепловых колебаний в гармоническом приближении можно рассчитать, проинтегрировав выражение для среднего значения энергии квантового осциллятора частоты ω по всему спектру частот, т.е. Z E= Ē (ω) g (ω) dω, (116) h̄ω h̄ω + h̄ω 2 e kT − 1 (117) ω где Ē (ω) = – средняя энергия осциллятора (см. п. 4.). Величина g (ω) представляет статистический вес системы. В теории Дебая (модель непрерывной упругой среды) для g (ω) используется представление g (ω) = 3V 2 ω , 2π2 v30 (118) Здесь V – объем системы, v0 – средняя скорость звуковых (упругих) волн в кристалле. Кроме того полагается, что верхний предел частот для кристаллических систем должен быть ограничен максимальной частотой (дебаевская частота), которая по порядку величины равна vd0 , где d – минимальное расстояние между частицами в решетке. Точно она определяется из соотношения ω Zmax g (ω) dω = 3N. 0 65 (119) Запишем выражение для полной энергии тепловых колебаний 3Vh̄ E = E0 + 2 3 2π v0 ω Zmax ω3 dω h̄ω 0 , e kT − 1 (120) где E0 – не зависящая от температуры энергия (нулевая). Для дальнейших исследований определим характеристическую температуру (температуру дебая) h̄ωmax TD = . (121) k Рассмотрим частные случаи выражение для E. Положим kT >> h̄ωmax . Тогда, предh̄ω ставляя e kT = 1 + h̄ω kT + ..., Получаем 3Vh̄ kT E(T ) = 2 3 · 2π v0 h̄ ω Zmax ω2 dω (122) 0 или E (T ) = 3NkT. (123) Теперь будем считать, что kT >> h̄ωmax . В этом случае верхний предел можно заменить на бесконечность и соответственно получаем E (T ) = 3π4 NkT 4 , 5TD (124) где использовано представление Z∞ x3 dx h̄4 = . ex − 1 15 (125) 0 Эти выражения позволяют определить молярную теплоёмкость кристаллической ре 3 4 шетки CV = 3R (T > TD ) и CV = 12π5 R TTD (T < TD ), где R – газовая постоянная. Значения TD (рассчитанные или полученные экспериментально) для простейших кристаллических структур находятся в интервале 200 − 400 К. Теплоёмкость кристаллической решётки практически полностью определять теплоёмкость диэлектриков и металлов. В последнем случае теплоёмкость валентных электронов не существенна, т.к. они являются вырожденной системой и их вклад определяется отношением kT µ0 << 1 для области реальных температур (см.п. 5.5). Подробное обсуждение вопроса о теплоёмкости твердых тел можно найти в специальной литературе: - Дж. Зейман, Принципы теории твёрдого тела, М.,1966., - Ч. Киттель, Введение в физику твёрдого тела, М.,1978, - А. Ансельм, Введение в физику полупроводников, М.,1962. Отметим, что тепловую энергию кристаллической решётки представляют также с помощью квазичастиц - фононов, понимая для возбуждённые состояния системы реальных частиц. Считают, что фононы относятся к классу бозе-частиц с химическим потенциалом равным нулю, массой покоя (m0 ) отличной от нуля и со скоростью "движения", равной скорости звука (v0 ). Таким образом, распределение для фотонов имеет вид 66 −1 N (ω) = eh̄ω/kT − 1 и расчёт термодинамических свойств формально можно осуществить также, как и для теплового равновесного излучения - фотонного газа (см.п. 5.7). Рассмотрим метод получения термического уравнения состояния кристаллической решётки. Будем представлять тепловое движение N частиц кристалла как 3N нормальных колебаний с максимальной частотой ωmax . Так как нормальные колебания не взаимодействуют друг с другом, то статистическая сумма для кристалла может быть представлена как произведение статистических сумм отдельных колебаний, т.е. Z = ∏ Zω i , (126) ωi где Zωi = e h̄ω − 2kTi 1−e − h̄ω kT - статистическая сумма квантового осциллятора частоты ωi (см.4). Сле- довательно свободная энергия F кристаллической решётки равна F = −kT · lnz = −kT ∑ lnzωi = ∑ Fωi , ωi ωi (127) h̄ω − kT где + kT ln 1 − e . Переходя к интегрированию получаем Fωi = −kT lnzωi = h̄ω 2 ω Zmax ω Zmax 0 0 Fω · g (ω) dω = F0 + kT F= h̄ω ln 1 − e− kT g (ω) dω Давление p определяется термодинамическим выражением ∂F p=− . ∂V T (128) (129) 6.4 Термодинамические свойства классической невырожденной плазмы Определим внутреннюю энергию E плазмы, считая что она представляет газовую полностью ионизированную среду. Энергия плазмы складывается из кинетической энергии теплового движения частиц ET и энергии электростатического взаимодействия Eq E = ET + Eq , (130) Если рассматривается классическая и невырожденная плазма, то энергия теплового движения на одну частицу (электроны, ионы) просто равна 32 kT . Получим выражение для энергии электростатического взаимодействия Eq , используя для неё общее представление Eq = 1 ∑ Niqiϕi. 2 i=1 (131) В данном случае будем полагать, что плазма состоит из заряженных частиц двух типов |+q| = |−q| = q и N0+ = N0− . В целом плазма электрически нейтральна. Для того, чтобы установить Eq , надо определить локальный потенциал . Это требует модельного представления плазмы. 67 Согласно предложению Дебая и Хюккеля, в системе свободных заряженных частиц, каждый заряд должен быть окружен пространственным облаком, в котором преобладают заряды противоположного знака (дебаевское экранирование). Исходя из такой модели определим локальный потенциал ϕi . Исходным в данном случае является уравнение Пуассона, примененное к каждому пространственному распределению заряда ∇2 ϕ = −4πρ, где ϕ (r) - результирующий потенциал электрического поля в распределении заряда, ρ (r) -пространственное распределение заряда. Предполагаем, что пространственные конфигурации обладают сферической симметрией. В данном случае получаем ∇2 ϕ = −4π ∑ qi ni , (132) i=1 где ni (r) – концентрация частиц i-ого типа. Для решения используем метод самосогласованного поля, т.к. будем считать, что qi ϕ ni = n0i e− kT , (133) где n0i – средняя концентрация по объёму. Уравнение Пуассона после подстановки ni (r) имеет вид для случая зарядов двух типов h i h i (+q)ϕ (−q)ϕ qϕ qϕ − − kT − 2 + − kT + − kT kT ∇ ϕ = −4π (+q) · n0 · e + (−q) · n0 · e = −4π qn0 e − qn0 · e , (134) где q – представляет величину заряда ±q. Будем считать, что qϕ << kT , т.е. имеет место слабое электростатическое взаимоqϕ qϕ действие. Тогда, используя для e± kT = 1 ± kT + ..., получаем + 2 2 n0 q n− 2 0q ∇ ϕ = 4π ϕ+ = χ2 ϕ. (135) kT kT 2 Здесь χ2 = 4π ∑ n0i q2i i=1 kT . Решение уравнения имеет вид ϕ (r) = qi −χr e . r (136) Искомое локальное поле определяется выражением h qi i ϕi = lim ϕ (r) − . r→0 r (137) При r → 0 имеем e−χr = 1 − χr + .... Тогда для ϕi получаем ϕi = −χqi . Определяем энергию электростатического взаимодействия π 1/2 1 1 Eq = ∑ Ni qi ϕi = − ∑ Ni q2i χ = − · 2 i 2 i kTV ∑ Niq2i !3/2 (138) i Дальнейший расчёт термодинамических величин осуществляется методами феноменологической термодинамики (см. И.П. Базаров, Термодинамика, М. Высшая школа, 1983). Свободная энергия Fq определяется выражением !3/2 r 2 π Fq = − Ni q2i . (139) 3 V kT ∑ i 68 Давление, связанное с электростатическим взаимодействием, имеет вид ∂Fq pq = − ∂V T 1 =− 32 3V / r π kT ∑ Niq2i !3/2 . (140) i Термодинамические свойства плазмы исследуют также и в случае вырожденного ее состояния. Такая ситуация, например, относится к электронному газу в поле положительной кристаллической решетки (металлы, полупроводники). Обсуждение такого вопроса можно найти в специальной литературе, например, Н.Ашкрофт, Н.Мермин, Физика твердого тела, М.,Мир ,1979. 6.5 Дополнительные вопросы 1. Показать, что для заданной конфигурации газа из N молекул вероятность обнаружить одну частицу в элементе объема d~r1 около~r1 , а другую частицу внутри элемента объема d~r2 около~r2 определяется выражением Z Z N (N − 1) EN f2 (~r1 ,~r2 ) d~r1 d~r2 = d~r1 d~r2 ... d~r3 ... d~rN exp − , (141) QN kT где EN – энергия взаимодействия N – частиц, QN – конфигурационный интеграл. Примечание. Считать, что состояние системы задается функцией канонического распределения. 2. Получить выражение для коэффициента A (см. п.6.2.) определяемого выражением Z E (r) 1 A (T ) = 1 − exp − d~r, (142) 2 kT где E(r) – радиальная потенциальная энергия взаимодействия двух частиц, для E(r) вида ∞, r ≤ d E (r) = (143) c rn , r > d, Здесь d – минимальное расстояние между частицами, например, равное d = 2R, где R - радиус частицы, c - положительная постоянная, n > 0. Примечание. Считать взаимодействие слабым E(r) << kT . Рассмотреть случаи n > 3 и n = 1. 3. Кристаллическая структура образована из N однотипных частиц, каждая из которых имеет три степени свободы. Будем представлять каждую частицу классическим гармоническим осциллятором. Покажите, что молярная теплоемкость такой системы равна Cv = 3R, где R – газовая постоянная. 4. Рассмотрим модель п.3. Однако, в данном случае считаем, что каждая частица системы представлена квантовым гармоническим осциллятором. Получите выражение для молярной теплоемкости системы. Обсудите результат для T < T = h̄ω k и для T > T, где T – характеристическая температура (по Эйнштейну). 5. Будем считать, что модель п.4. состоит из различных частиц так, что каждому осциллятору соответствует частота ωi , где 1 ≤ i ≤ N. Можно ли ожидать, что в этом случае для теплоемкости будут получены результаты, соответствующие модели Дебая (см. п. 6.3.). 6. Запишите выражение для полной энергии равновесной газовой плазмы, учитывая, что такое состояние среды достигается при T ≈ 105 K. 69 7. Используя частные условия устойчивости термодинамических систем (см. п.1.), покажите, что при одинаковых условиях плазма менее устойчива, чем идеальный газ таких же частиц. 8. Дополнение к п.6.4. а) Доказать, что плотность числа отрицательно заряженных частиц в присутствии дополнительного точечного заряда q (положительного) определяется выражением n (r) = n0 + qχ2 ϕ (r) . 4πqe (144) б) Определить полное число избыточных отрицательно заряженных частиц внутри сферы Дебая. в) Найти энергию взаимодействия между зарядом q и зарядом в сфере Дебая. г) Найти энергию распределенного отрицательного заряда внутри сферы Дебая. 7 Равновесные флуктуации 7.1 Флуктуации энергии Рассматривается закрытая система, состояние которой представляется каноническим распределением (п.2.3.). Получим выражение для среднеквадратичной флуктуации (дисперсии) энергии такой системы. По определению ∆E 2 = E 2 − Ē 2 . (145) Определим выражения для слагаемых правой части 1 En En Ē = ∑ En ωn = ∑ En exp − , Z = ∑ exp − Z n kT kT n n или 1 ∂Z , Z ∂β 1 ; kT 1 En 2 2 2 E = ∑ En ωn = ∑ En exp − Z n kT n Ē = − или E2 = β= 1 ∂2 Z . Z ∂β2 (146) (147) (148) (149) Таким образом имеем ∆E 2 1 ∂2 Z 1 = − 2 2 Z ∂β Z ∂Z ∂β 2 . (150) В свою очередь ∂Ē ∂ 1 ∂Z 1 ∂2 Z 1 ∂Z 2 = − =− + . ∂β ∂β Z ∂β Z ∂β2 Z 2 ∂β Отсюда получаем ∆E 2 = − где для макроскопической системы ∂Ē ∂β ∂Ē ∂T ∆E 2 = kT 2 = CV . 70 ∂Ē , ∂T (151) (152) 7.2 Флуктуации числа частиц Рассматривается открытая система, состояние которой представляется обобщенным распределением (п.2.4.). Получим выражение для среднеквадратичной флуктуации (дисперсии) числа частиц в системе. По определению ∆N 2 = N 2 − N̄ 2 . (153) Определим выражения для слагаемых в правой части µNn − En,N 1 N̄ = ∑ ∑ Nn ωn,N = , ∑ Nnexp Z0 ∑ kT N n N n µNn −En,N где Z0 = ∑ ∑ Nn exp . kT (154) N n Отсюда следует N̄ = kT ∂Z0 · . Z0 ∂µ (155) Выражение для N 2 имеет вид N2 = ∑∑ µNn − En,N 1 2 = ∑ Nn exp Z0 ∑ kT N n Nn2 ωn,N N n или N2 (156) (kT )2 ∂2 Z0 = · 2 . Z0 ∂µ (157) Таким образом получаем " ∆N 2 = (kT ) 2 1 ∂ 2 Z0 1 − 2 2 Z0 ∂µ Z0 ∂Z0 ∂µ 2 # (158) . В свою очередь ∂N̄ ∂ = ∂µ ∂µ kT ∂Z0 · Z0 ∂µ " 1 ∂ 2 Z0 1 = kT · 2 − 2 Z0 ∂µ Z0 Отсюда следует ∆N 2 = kT ∂Z0 ∂µ 2 # . ∂N̄ . ∂µ (159) (160) Естественно формулы для ∆E 2 и ∆N 2 применимы и для классических систем. 7.3 Эквивалентность функций распределений Гиббса Флуктуации энергии и числа частиц в термодинамических системах (N → ∞, V → ∞) несущественны. Для демонстрации этого факта рассмотрим примеры. Для классического идеального газа бесструктурных частиц Ē = N 32 kT . Тогда получаем для дисперсии ∆E 2 = 32 N (kT )2 , а относительная флуктуация равна p ∆E 2 1 δE = ≈√ (161) Ē N 71 Для классического идеального газа бесструктурных частиц (2πmkT )3/2 V µ N̄ = exp . 3 h kT (162) Тогда для дисперсии ∆N 2 имеем ∆N 2 = N̄, а относительная флуктуация равна p ∆N 2 1 ≈√ δE = N̄ N (163) В связи с этим следует считать, что классификация термодинамических систем на изолированную, закрытую и открытую (см. п. 1.1.) и применение к ним соответствующих функций распределения является достаточно условным. Такие представления только расширяют возможности математического аппарата статистической термодинамики. Состояние любой термодинамической системы можно представлять любой из трех рассмотренных (п.2.) функций распределения Гиббса в зависимости от условий рассматриваемой проблемы. В этом смысле они являются эквивалентными функциями распределения. 7.4 Распределение вероятности для флуктуаций Допустим, функция распределения f (A) определяет значение некоторой величины A, представляющей термодинамическую систему и которая испытывает флуктуации, т.е. является случайной величиной. Функция f (A) нормирована Z (164) f (A) dA = 1. Определим функцию f (A) в виде 5 f (A) = C · eS(A) , C= R 1 eS(A) dA . (165) Здесь S(A) – энтропия термодинамической системы в состоянии с данным значением A. Учитывая малость флуктуаций, можно разложить энтропию S(A) в ряд по степеням A относительно ее максимального значения, соответствующего равновесному состоянию системы. dS 1 d2S S (A) = S0 + A+ + ... , (166) dA 0 2 dA2 0 где индекс "О"обозначает равновесное состояние системы. В состоянии равновесия выполняется (см. п. 1..) 2 dS d S =0 < 0. (167) dA 0 dA2 0 Тогда для функции распределения f (A) получаем −σA2 f (A) = C · e 1 σ=− 2 , d2S dA2 > 0. (168) 0 5 Обоснование этого выражения приводится в литературе: А.И. Ансельм "Основы статистической фи- зики и термодинамики", М., Наука, 1983.; Ю.Л. Климантович "Статистическая физика", М., Наука, 1983. 72 Постоянную C в данном выражении определяем из условия нормировки Z+∞ Z+∞ f (A) dA = C −∞ −σA2 e r dA = C −∞ π = 1. σ (169) Среднеквадратичное значение A2 определяется как A2 = Z+∞ r 2 A f (A) dA = −∞ Таким образом получаем, что σ = σ π Z+∞ 2 −σA2 A e r dA = −∞ 1 . 2A2 √ 1 σ1 π = . π 2 σ3/2 2σ (170) Эти выражения позволяют записать 1 f (A) = p − e A2 2A2 (171) , 2πA2 что соответствует центрированному распределению Гаусса (Ā = 0). 7.5 Флуктуация основных термодинамических величин Будем считать, что энтропия S системы есть функция двух переменных энергии E и объема V , т.е. S = S(E,V ). Изменения S связаны с флуктуациями E и V . Осуществим разложение S в ряд Тейлора для функции двух переменных S (E,V ) = S (E0 ,V0 ) + [SE0 (E0 ,V0 ) ∆E + SV0 (E0 ,V0 ) ∆V ] + 00 (E ,V ) ∆E∆V + S00 (E ,V ) ∆V 2 + ... + 12 SE00 (E0 ,V0 ) ∆E 2 + 2SEV 0 0 0 0 V (172) Первые производные SE0 SV0 равны нулю вследствие равновесного состояния системы. Рассмотрим второе слагаемое 1 ∂2 S 2 ∂2 S ∂2 S 2 ∆p∆V − ∆T ∆S ∆E + 2 ∆E∆V + 2 ∆V = , (173) 2 2 ∂E ∂E∂V ∂V 2T где в процессе преобразований использовано основное термодинамическое равенство dE = T dS − pdV . Рассмотрим общее выражение для функции распределения (см.п.7.4.) в данном случае 6 ∆p∆V − ∆T ∆S f (p,V, T, S) = C · exp . (174) 2kT Это выражение позволяет получить информацию о флуктуациях основных термодинамических величин. Выберем в качестве пары независимых переменных V и T . Тогда имеем ∂p ∂p ∆p = ∆V + ∆T , (175) ∂V T ∂T V ∆S = 6В ∂S ∂V ∂S ∆V + ∂T T ∆T = V ∂p ∂T V ∆V + CV ∆T. T показателе экспоненты введена постоянная Больцмана k для выполнения размерности. 73 (176) В этом случае для f (V, T ) имеем 1 f (V, T ) = Cexp 2kT ∂p ∂V CV 2 ∆V − ∆T . 2kT 2 T 2 (177) Отсюда получаем ∂p 1 ∆V 2 h 2kT ∂V Ti CV 2 . (T ) = C2 exp − 2kT 2 ∆T f (V ) = C1 exp f h i , (178) Имея в виду гауссовый характер распределения флуктуаций получаем для дисперсий следующие выражения ∂V kT 2 ∆V 2 = −kT , ∆T 2 = (179) ∂p T CV и кроме того ∆V · ∆T = 0. Для определения флуктуаций других величин выбирают другие пары независимых переменных. Например, если взять P и S то получим 1 ∂V 1 2 2 f (P, S) = Cexp ∆p − ∆S . (180) 2kT ∂p S 2kC p Отсюда получаем ∆p2 = − kT , ∂V ∂p S ∆S2 = kC p , ∆p · ∆S = 0. (181) 7.6 Дополнительные вопросы 1. Для распределения Максвелла по скоростям получить следующие величины: а) Среднее значение n-ой степени скорости 2 Vn = √ π 2kT m n/2 n+3 Γ , 2 где n - вещественное число, n>-1, Г - гамма-функция. б) Дисперсию скорости kT 8 V2 = 3− . m π (182) (183) в) Дисперсию кинетической энергии 1 m 2 2 v2 − v2 2 = 3 (kT )2 . 2 (184) г) Для п. б) и в) получить выражение для относительных флуктуаций этих величин. 2. Показать, что для любой макроскопической системы, состоящей из невзаимодействующих однотипных подсистем, число которых равно N, выполняется соотношение для дисперсии энергии ∆EN2 = N∆E 2 , где ∆E 2 – дисперсия энергии подсистемы. 3. Применяя результат п.7.1. найти среднеквадратичную флуктуацию и относительную среднеквадратичную флуктуацию а)гармонического осциллятора (считать энергию 74 основного состояния равной нулю), б)набора из N одинаковых гармонических осцилля2 0 торов. Для а) и б) рассмотреть случай kT >> hν0 . Ответ: а) ∆E = exph̄ω kT ; при kT >> hν0 Ē 2 ∆E 2 = (kT )2 , ∆E = 1. Ē 2 2 ∆E h̄ω 1 N б) ∆EN2 = N∆E12 ; = N 1 + Ē0 . 2 2 получаем Ē = kT, EN 4. Применяя модель Дебая (п.5.3.), найти температурную зависимость относительных флуктуаций колебательной энергии кристалла (V = Const) в случае низких температур. Ответ: ∆E 2 Ē 2 2 = kTĒ 2CV = NA - число Авогадро). 16 12 4 5 π N T 3 T , где CV 4 = 12 5π R 3 T T 4 4 T , Ē = 12 5 π R 4T 3 , R - постоянная (R = NA k, 5. Рассматривается система, представляемая обобщенным распределением ωn,N = µNn − En,N 1 exp . Z0 kT (185) Показать, что в этом случае справедливы выражения ∆E∆N = kT ∂E , ∂µ T ∂E ∆E 2 = kT 2 ∂E + kT µ . ∂T ∂µ µ (186) T Решение. Первый результат получают при дифференцировании Ē по ??? при T = Const. Второй результат получают дифференцированием Ē по T при ? = Const с учетом результата а). 6. Используя результат п.7.2. найти дисперсию числа заполнения для одночастичного состояния в случаях, когда частицы подчиняются статистике Ферми-Дирака и БозеЭйнштейна. Ответ: ∆N 2 = N̄ − N 2 ; ∆N 2 = N̄ + N 2 . 75